Agencia Estatal Boletín Oficial del Estado






Como consecuencia de la adhesión de España a las Comunidades Europeas y recogiendo los principios de la Directiva 71/307/CEE del Consejo, de 26 de julio de 1971, relativa a la aproximación de las legislaciones de los Estados miembros sobre las denominaciones textiles, y sus modificaciones posteriores, se aprobó el Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.
Dado que para el cumplimiento del citado real decreto era necesario establecer unos métodos de análisis cuantitativos de mezclas binarias y ternarias de fibras textiles, adaptando así nuestra normativa a las exigencias de la legislación comunitaria, por medio del Real Decreto 656/1989, de 2 de junio, por el que se aprueban los métodos de análisis cuantitativos de mezclas binarias y terciarias de fibras textiles, se declararon oficiales los métodos establecidos por la Comunidad Europea mediante las Directivas 72/276/CEE y 73/44/CEE del Consejo, de 17 de julio de 1972 y de 26 de febrero de 1973, respectivamente, y sus posteriores modificaciones, con objeto de que la determinación de la composición en fibras de los productos textiles, tanto en lo que se refiere al tratamiento previo de la muestra como a su análisis cuantitativo, se efectuase a través de métodos uniformes.
Por otra parte, en el año 1996 se aprobó la Directiva 96/74/CE del Parlamento Europeo y del Consejo, de 16 de diciembre de 1996, relativa a las denominaciones textiles, por la que se prescribe un etiquetado que indique la composición de la fibra de los productos textiles, así como la realización de pruebas mediante análisis de la conformidad de dichos productos con las indicaciones que figuren en su etiqueta.
Como consecuencia de ello, ese mismo año se aprobó también la Directiva 96/73/CE del Parlamento Europeo y del Consejo, de 16 de diciembre de 1996, sobre determinados métodos de análisis cuantitativos de mezclas binarias de fibras textiles, que estableció métodos uniformes de análisis cuantitativos de mezclas binarias de fibras textiles.
Posteriormente, la Directiva 97/37/CE de la Comisión, de 19 de junio de 1997, llevó a cabo una adaptación al progreso técnico de los anexos I y II de la Directiva 96/74/CE. Dicha Directiva 96/73/CE se incorporó al ordenamiento interno por el Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.
También, con la misma finalidad de adaptación al progreso técnico, se aprobó la Directiva 2004/34/CE de la Comisión, de 23 de marzo de 2004, por la que se modifican, con objeto de adaptarlos al progreso técnico, los anexos I y II de la Directiva 96/74/CE, que fue incorporada al ordenamiento interno por el Real Decreto 2322/2004, de 17 de diciembre, por el que se añade la fibra polilactida a los anexos I y II del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.
Asimismo, se aprobó la Directiva 2006/3/CE de la Comisión, de 9 de enero de 2006, por la que se modifican, para su adaptación al progreso técnico, los anexos I y II de la Directiva 96/74/CE, con objeto de añadir el elastomultiéster a la lista de fibras que figura en sus anexos I y II, cuya incorporación al ordenamiento interno se ha realizado por medio de Real Decreto 1115/2006, de 29 de septiembre, por el que se modifica el Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.
En función de ello, la Comisión Europea ha considerado que era necesario definir métodos uniformes de ensayo para la polilactida y el elastomultiéster. A tal efecto se ha aprobado la Directiva 2006/2/CE de la Comisión, de 6 de enero de 2006, por la que se modifica, para su adaptación al progreso técnico, el anexo II de la Directiva 96/73/CE del Parlamento Europeo y del Consejo sobre determinados métodos de análisis cuantitativos de mezclas binarias de fibras textiles.
En virtud de esta circunstancia y en aras de una mayor racionalidad y claridad respecto a los métodos de análisis cuantitativos de mezclas binarias de fibras textiles, por medio de este real decreto se procede a la incorporación al ordenamiento jurídico interno de las Directivas 96/73/CE del Parlamento Europeo y del Consejo, de 16 de diciembre de 1996, y 2006/2/CE de la Comisión, de 6 de enero de 2006.
Este real decreto se dicta al amparo del artículo 149.1.13.a de la Constitución, que atribuye al Estado competencia exclusiva en materia de bases y coordinación de la planificación general de la actividad económica.
En la tramitación de este real decreto se ha dado audiencia a las asociaciones de consumidores y usuarios y a los sectores afectados, habiéndose realizado igualmente un trámite de consulta a las comunidades autónomas.
En su virtud, a propuesta de los Ministros de Sanidad y Consumo y de Industria, Turismo y Comercio, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión del día 12 de enero de 2007,
DISPONGO:
1. Este real decreto establece los métodos oficiales de análisis cuantitativos de mezclas binarias de fibras textiles y el modelo de preparación de las muestras reducidas y de las muestras de análisis.
2. A los efectos del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles, se declaran de obligado cumplimiento los citados métodos de análisis.
1. Por muestra reducida se entiende una muestra de un tamaño apropiado para los análisis, procedente de muestras globales para laboratorio tomadas a su vez de una partida de artículos para analizar.
2. Por muestra de análisis se entiende la porción de la muestra reducida necesaria para obtener un resultado analítico individual.
En los controles oficiales que se efectúen para determinar la composición de productos textiles ya comercializados, se aplicarán las disposiciones de los anexos I y II relativas al modo de preparación de muestras reducidas y muestras de análisis y a los métodos de análisis cuantitativos de determinadas mezclas binarias de fibras textiles.
El laboratorio encargado del control de las mezclas binarias para las que no exista método de análisis uniforme en el ámbito comunitario, determinará la composición de estas mezclas utilizando cualquier método válido a su disposición, e indicará en el informe del análisis el resultado obtenido y la precisión del método, si fuese conocida.
Sin perjuicio de la potestad sancionadora del Ministerio de Industria, Turismo y Comercio, en el ejercicio de sus propias competencias, las actuaciones administrativas en materia de defensa del consumidor, en cuanto a la toma de muestras y análisis de los productos textiles se refiere, se regirán por lo dispuesto en el Real Decreto 1945/1983, de 22 de junio, por el que se regulan las infracciones y sanciones en materia de defensa del consumidor y de la producción agroalimentaria.
De acuerdo con el artículo anterior, la recogida de muestras oficiales de carácter global para la realización de análisis, se formalizará por triplicado según lo previsto en el artículo 15 del Real Decreto 1945/1983, de 22 de junio.
A tales efectos, cada muestra constará de una cantidad de producto que, al menos, tenga un peso de 50 gramos. En artículos de peso superior a los 150 gramos, los servicios oficiales de inspección podrán constituir cada muestra por partición del artículo, procurando que cada una de ellas sea lo más homogénea posible en relación al producto total.
Para artículos con menor peso al descrito, se procederá a las tomas de tres unidades iguales.
Por medio de este real decreto se derogan las disposiciones del Real Decreto 656/1989, de 2 de junio, por el que se aprueban los métodos de análisis cuantitativos de mezclas binarias y terciarias de fibras textiles, en lo que se refiere a las mezclas binarias de fibras textiles, manteniendo su vigencia en cuanto a las mezclas ternarias de dichas fibras.
Se modifica el título del Real Decreto 656/1989, de 2 de junio, por el que se aprueban los métodos de análisis cuantitativos de mezclas binarias y terciarias de fibras textiles que pasa a denominarse Real Decreto 656/1989, de 2 de junio, por el que se aprueban los métodos de análisis cuantitativos de mezclas ternarias de fibras textiles.
Este real decreto se dicta al amparo del artículo 149.1.13.a de la Constitución, que atribuye al Estado competencia exclusiva en materia de bases y coordinación de la planificación general de la actividad económica.
Mediante este real decreto se incorporan al ordenamiento jurídico interno las Directivas 96/73/CE del Parlamento Europeo y del Consejo, de 16 de diciembre de 1996, y 2006/2/CE de la Comisión, de 6 de enero de 2006, por la que se modifica, para su adaptación al progreso técnico, el anexo II de la Directiva 96/73/CE.
El presente real decreto entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
Dado en Madrid, el 12 de enero de 2007.
JUAN CARLOS R.
La Vicepresidenta Primera del Gobierno y Ministra de la Presidencia,
MARÍA TERESA FERNÁNDEZ DE LA VEGA SANZ
El presente Anexo tiene por objeto fijar el procedimiento que deberá seguirse para la preparación de muestras reducidas de un tamaño apropiado (es decir, de un peso no superior a 100 g) para los tratamientos previos a los análisis cuantitativos a partir de muestras globales para laboratorio, y para la selección de muestras de análisis a partir de muestras reducidas que hayan sido sometidas a tratamiento previo para eliminar las materias no fibrosas1.
1 En ciertos casos, las muestras de análisis podrán someterse directamente a tratamiento previo.
2.1. Partida. Es la cantidad de material que se valora sobre la base de una serie de resultados de pruebas. Puede comprender, por ejemplo, todo el material que corresponda a una misma entrega de tejido, toda la tela tejida a partir de un enjulio determinado, una expedición de hilados, una o varias balas de fibras en bruto.
2.2. Muestra global para laboratorio. Es aquella porción del lote tomada de forma que sea representativa del conjunto, y que se envía al laboratorio. La muestra global para laboratorio tendrá el tamaño y la naturaleza que basten para reflejar convenientemente la variabilidad del lote y para que sea fácil su manipulación en el laboratorio2.
2 Para los artículos acabados y confeccionados véase el punto 7.
2.3. Muestra reducida. Es la porción de la muestra global para laboratorio que se somete a un tratamiento previo para eliminar las materias no fibrosas, y de la cual se toman las muestras para el análisis. La muestra reducida tendrá el tamaño y la naturaleza que basten para reflejar convenientemente la variabilidad de la muestra global para laboratorio3.
3 Véase el punto 1.
2.4. Muestra de análisis o toma de prueba. Es la porción de material tomada de la muestra reducida necesaria para dar un resultado analítico individual.
La muestra reducida se escogerá de manera que sea representativa de la muestra global para laboratorio.
Las muestras de análisis se tomarán de la muestra reducida de manera que sean representativas de esta última.
4.1. Fibras no orientadas. Constituir la muestra reducida tomando al azar varias mechas de la muestra global para laboratorio. Mezclar convenientemente toda la muestra reducida con ayuda de una carda de laboratorio4. Someter el velo o la mezcla así obtenidos a tratamiento previo, incluyendo las fibras sueltas y las que se adhieran al aparato utilizado para la mezcla. Tomar a continuación muestras de análisis del velo, de las fibras adherentes y de las que se deslicen fuera del aparato, en proporción al peso.
4 Se podrá sustituir la carda de laboratorio por un mezclador de fibras o por el método llamado «enjambres de mechones».
Si el velo de carda permaneciera intacto después del tratamiento previo, tomar las muestras de análisis del modo descrito en el número 4.2. Si el velo hubiese sido afectado por el tratamiento previo, escoger en él las muestras tomando al azar un mínimo de 15 mechas pequeñas de tamaño apropiado y aproximadamente iguales, reuniéndolas a continuación.
4.2. Fibras orientadas (velos de carda, cintas, mechas). De partes de la muestra global para laboratorio escogidas al azar cortar un mínimo de 10 secciones transversales que pesen cada una alrededor de 1 g.
Someter la muestra reducida así formada a tratamiento previo. Reunir a continuación las secciones colocándolas una al lado de la otra y formar la muestra de análisis cortando transversalmente de manera que se tome una porción de cada una de las 10 longitudes.
5.1. Hilos en bobinas o en madejas. Tomar muestras de todas las bobinas de la muestra global para laboratorio.
Retirar de cada bobina longitudes continuas, iguales y apropiadas, sea devanando ovillos de un mismo número de vueltas en una devanadera5 o por cualquier otro medio. Reunir los largos uno al lado de otro, en forma de ovillo único o cable y cuidando que en el ovillo o cable haya largos iguales de cada bobina.
5 Si las bobinas pudieran ponerse en un portabobinas apropiado, podría desenrollarse simultáneamente un gran número de ellas.
Someter la muestra reducida así formada a tratamiento previo.
Tomar las muestras de análisis de la muestra reducida cortando del ovillo o del cable un haz de hilos de igual longitud, teniendo cuidado que el haz contenga todos los hilos de la muestra.
Si t es el «tex» del hilo, y n el número de bobinas de la muestra global para laboratorio, habrá que sacar de cada bobina una longitud de hilo de longitud de hilo de 106/nt para obtener una muestra reducida de 10 g.
Si nt es alto, es decir, si es superior a 2000, podrá tejerse un ovillo de mayor peso y cortarlo transversalmente en dos puntos, de modo que se obtenga un cable de un peso apropiado. Las extremidades de una muestra que se presente en forma de cable se atarán convenientemente antes de someterlas al tratamiento previo, tomándose las muestras de análisis a suficiente distancia del nudo.
5.2. Hilo sobre enjulio. Tomar una muestra reducida cortando en la extremidad del enjulio un haz de al menos 20 cm. de largo que contenga todos los hilos, a excepción de los hilos de la orilla, que se desecharán. Atar el haz de hilos por una de sus extremidades. Si la muestra fuera demasiado ancha para someterla entera a tratamiento previo, dividirla en dos o más partes, atar cada una de ellas por separado, someterlas a tratamiento previo también por separado y reunirlas de nuevo una vez concluido éste. Tomar una muestra de análisis de longitud conveniente de la muestra reducida, cortándola a suficiente distancia del nudo y sin dejarse ninguno de los hilos del enjulio. Para enjulios que contengan N hilos de t «tex», la longitud de una muestra que pese 1 g será de 105/nt cm.
6.1. Muestra global para laboratorio constituida por un retazo único representativo del tejido. Cortar en la muestra una banda diagonal que vaya de una esquina a otra y quitar las orillas. Esta banda constituirá la muestra reducida. Para obtener una muestra reducida de x g, la superficie de la banda será de x104/G cm2, siendo G el peso del tejido en g por m2.
Después de haberla sometido al tratamiento previo, cortar la banda transversalmente en cuatro partes iguales y ponerlas unas encima de otras. Tomar las muestras de análisis de una parte cualquiera del material así preparado, cortando transversalmente todas las capas, de manera que cada muestra comprenda una longitud igual de cada una de ellas.
Si la tela presentase un dibujo tejido, la anchura de la muestra reducida, medida paralelamente a la dirección de la urdimbre, no deberá ser inferior a la distancia que exista entre repeticiones del dibujo en la urdimbre. Si, cumplida esta condición, la muestra reducida fuese demasiado ancha para ser sometida entera a tratamiento previo, córtese en partes iguales, sométanse éstas a tratamiento previo separadamente y colóquense unas encima de otras antes de tomar las pruebas de análisis, tratando que las partes correspondientes del dibujo no coincidan.
6.2. Muestra global para laboratorio constituida por varios retazos: tratar cada retazo según lo dispuesto en el número 6.1 y dar los resultados por separado.
La muestra global para laboratorio estará normalmente constituida por un artículo acabado y confeccionado, o por una fracción representativa de uno de estos artículos.
Determinar en su caso el porcentaje de las diferentes partes del artículo que no tengan el mismo contenido en fibras, con objeto de comprobar si se cumplen las disposiciones del artículo 6 del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.
Tomar una muestra reducida representativa de la parte del artículo acabado y confeccionado, cuya composición deberá indicarse en la etiqueta. Si el artículo confeccionado llevara varias etiquetas, deberán tomarse muestras reducidas representativas de la parte o partes correspondientes a cada etiqueta.
Si el artículo cuya composición se trata de determinar no fuese homogéneo, pudiera ser necesario tomar muestras reducidas de cada una de las partes del artículo y determinar las proporciones relativas de las diversas partes con relación al conjunto del artículo de que se trate.
El cálculo de los porcentajes se hará teniendo en cuenta las proporciones relativas de las partes de las que se tomen muestras.
Someter las muestras reducidas a tratamiento previo.
Tomar a continuación muestras de análisis representativas de las muestras reducidas sometidas a tratamiento previo.
1. GENERALIDADES
Introducción.
Los métodos de análisis cuantitativo de las mezclas de fibras textiles se basan en dos procedimientos principales, el de la separación manual y el de la separación química de las fibras.
El procedimiento de separación manual deberá utilizarse siempre que sea posible, ya que generalmente se obtienen con él resultados más precisos que con el procedimiento químico. Es aplicable a todos los productos textiles en los que las fibras componentes no formen una mezcla íntima, como por ejemplo en el caso de hilados compuestos de varios elementos cada uno de ellos constituido por una sola clase de fibra, o de tejidos en los que la fibra que compone la urdimbre sea de naturaleza diferente a la que compone la trama, o de géneros de punto que puedan destejerse compuestos de hilos de diversas clases.
El procedimiento de análisis químico cuantitativo de mezclas de fibras textiles se funda generalmente en la solubilidad selectiva de los componentes individuales de la mezcla. Después de la eliminación de uno de los componentes, el residuo insoluble se pesa y la proporción del componente soluble se calcula a partir de la pérdida de peso. Esta primera parte del Anexo contiene la información común a los análisis por este procedimiento de todas las mezclas de fibras consideradas en el presente Anexo, cualquiera que sea su composición. Deberá por tanto utilizarse conjuntamente con aquellas secciones del Anexo que contengan los procedimientos detallados aplicables a mezclas de fibras concretas. Puede ocurrir que algunos análisis químicos se basen en un principio que no sea el de la solubilidad selectiva. En estos casos, podrá hallarse información completa y detallada sobre ello en la sección correspondiente del método aplicable.
Las mezclas de fibras utilizadas durante la fabricación de los productos textiles y, en menor grado, las que se encuentran en los productos acabados, contienen a veces materias no fibrosas como grasas, ceras o aditivos, o productos solubles en el agua, que pueden tener origen natural o haber sido añadidos para facilitar la fabricación. Las materias no fibrosas deberán eliminarse antes del análisis. Ésta es la razón por la cual se describe igualmente un método de tratamiento previo que permite eliminar los aceites, las grasas, las ceras y los productos solubles en el agua en la mayoría de los casos.
Los productos textiles pueden contener además resinas u otras materias añadidas para conferirles propiedades especiales. Tales materias, incluidos los colorantes en ciertos casos excepcionales, pueden modificar la acción del reactivo sobre el componente soluble e incluso ser parcial o totalmente eliminadas por los reactivos. Estas materias añadidas pueden por tanto inducir a error y deberán eliminarse antes de analizar la muestra. En caso de que esta eliminación sea imposible, los métodos de análisis químico cuantitativo descritos en el presente Anexo no serán aplicables.
El colorante presente en las fibras teñidas se considera como parte integrante de la fibra y no se eliminará.
Estos análisis se efectúan sobre la base del peso en seco y se suministra un método para determinarlo.
El resultado se obtendrá aplicando al peso en seco de cada fibra los porcentajes convencionales indicados en el Anexo II del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.
Las fibras presentes en la mezcla deberán identificarse antes de efectuar los análisis. En ciertos métodos químicos, la parte insoluble de los componentes de una mezcla podrá disolverse parcialmente en el reactivo utilizado para disolver el componente soluble. Siempre que sea posible, se escogerán reactivos que tengan un efecto débil o nulo sobre las fibras insolubles. Si se supiera que durante el análisis se produce una pérdida de peso, convendrá corregir el resultado; se acompañan factores de corrección a tal fin. Estos factores han sido determinados en diferentes laboratorios tratando las fibras depuradas mediante tratamiento previo con el reactivo apropiado especificado en el método de análisis. Estos factores sólo se aplicarán a fibras normales y podrán ser necesarios otros factores de corrección si las fibras se hubiesen deteriorado antes o durante el tratamiento. Los métodos químicos propuestos se aplicarán a análisis individuales. Convendrá efectuar como mínimo dos análisis sobre muestras de análisis separadas, tanto cuando se siga el procedimiento de separación manual como cuando se utilice el de separación química. En caso de duda, y salvo imposibilidad técnica, se deberá efectuar otro análisis siguiendo un método alternativo que permita la disolución de las fibras no disueltas al utilizar el primer método.
Informaciones comunes a los métodos que deberán seguirse, para el análisis químico cuantitativo de mezclas de fibras textiles.
I.1. Ámbito de aplicación.
En el ámbito de aplicación de cada método se indican las fibras a las que el método es aplicable.
I.2. Principio.
Después de haber identificado los componentes de una mezcla, se eliminarán en primer lugar las materias no fibrosas por medio de un tratamiento previo apropiado y después uno de los dos componentes, generalmente por disolución selectiva6. Se pesará el residuo insoluble y se calculará la proporción del componente soluble a partir de la pérdida de peso. Salvo que ello plantee dificultades técnicas, será preferible disolver la fibra presente en mayor proporción, a fin de obtener como residuo la fibra que se encuentre en menor proporción.
6 El método n° 12 constituye una excepción. Se basa en la determinación del contenido en un elemento constitutivo de uno de los componentes.
I.3. Material necesario.
I.3.1. Instrumental.
I.3.1.1. Placas filtrantes y pesafiltros que permitan la incorporación de placas, o cualquier otro instrumental que dé idénticos resultados.
I.3.1.2. Matraz de succión.
I.3.1.3. Desecador que contenga gel de sílice coloreado mediante un indicador.
I.3.1.4 Horno de secado con ventilador para secar las muestras a 105 ºC ± 3 ºC.
I.3.1.5. Balanza analítica, sensibilidad 0,0002 g.
I.3.1.6. Equipo de extracción Soxhlet o instrumental que permita alcanzar idénticos resultados.
I.3.2. Reactivos.
I.3.2.1. Éter de petróleo redestilado, con punto de ebullición entre 40 ºC y 60 ºC.
I.3.2.2. Los otros reactivos se mencionan en la sección correspondiente de cada método. Todos los reactivos utilizados deberán ser químicamente puros.
I.3.2.3. Agua destilada o desionizada.
I.4. Atmósfera de acondicionamiento y de análisis.
Como lo que se determina es el peso en seco, no será necesario acondicionar las muestras ni hacer los análisis en una atmósfera acondicionada.
I.5. Muestra reducida.
Escoger una muestra reducida representativa de la muestra global para laboratorio suficiente para suministrar todas las muestras de análisis necesarias de 1 g como mínimo cada una.
I.6. Tratamiento previo de la muestra reducida7.
7 Ver Anexo I.1
Si estuviese presente un elemento que no deba tenerse en cuenta para el cálculo de los porcentajes (ver el apartado 3 del artículo 7 del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles), se comenzará por eliminarlo mediante un método apropiado que no afecte a ninguno de los componentes fibrosos.
Con esta finalidad, las materias no fibrosas que se puedan extraer con éter de petróleo y agua se eliminarán tratando la muestra reducida, secada al aire, en el extractor Soxhlet con éter de petróleo durante una hora, a una cadencia mínima de seis ciclos por hora. Evaporar el éter de petróleo de la muestra, que se extraerá después por tratamiento directo mediante inmersión durante una hora en agua a temperatura ambiente, seguida de inmersión durante una hora en agua a 65 ºC ± 5 ºC, agitando de vez en cuando. La relación muestra/agua será de 1/100. Eliminar el exceso de agua de la muestra por estrujamiento, succión o centrifugación. A continuación, dejar secar la muestra al aire.
En caso de que las materias no fibrosas no pudieran extraerse con éter de petróleo y agua, deberán eliminarse sustituyendo el procedimiento descrito más arriba por un procedimiento apropiado que no altere sustancialmente ninguno de los componentes fibrosos. Sin embargo, para ciertas fibras vegetales naturales crudas (yute, coco, por ejemplo) hay que señalar que el tratamiento previo normal con éter de petróleo y agua no elimina todas las sustancias no fibrosas naturales; a pesar de ello, no se aplicarán tratamientos previos complementarios a menos que la muestra contenga aprestos no solubles en el éter de petróleo y en el agua.
En los informes del análisis deberán describirse detalladamente los métodos de tratamiento previo utilizados.
I.7. Procedimiento de análisis.
I.7.1. Instrucciones generales.
I.7.1.1. Secado.
Todas las operaciones de secado deberán efectuarse en un tiempo no inferior a 4 horas ni superior a 16 horas, a 105 ºC ± 3 ºC, en un horno ventilado cuya puerta permanezca cerrada durante toda la duración del secado. Si la duración del secado fuera inferior a 14 horas, deberá comprobarse si se ha obtenido una masa constante. Se considerará alcanzada ésta cuando la variación de masa, después de un nuevo secado de 60 minutos, sea inferior a 0,05 %.
Durante las operaciones de secado, de enfriamiento y de pesado, evitar manipular las placas filtrantes y los pesafiltros, las tomas de pruebas o los residuos con las manos desnudas.
Secar las muestras en un vidrio de reloj sin taparlo, pero con la tapa metida también en el horno. Después del secado, tapar el pesafiltros antes de sacarlo del horno y ponerlo rápidamente en el desecador.
Secar en el horno la placa filtrante colocada en un vidrio de reloj destapado. Introducir también en el horno la tapa de éste. Después del secado, tapar el pesafiltros y ponerlo rápidamente en el desecador.
En caso de que se emplease un instrumental que no fuese la placa filtrante, las operaciones de secado en el horno se llevarán a cabo de manera que el peso en seco de las fibras pueda determinarse sin pérdida.
I.7.1.2. Enfriamiento.
Efectuar todas las operaciones de enfriamiento en el desecador, manteniéndolo al lado de la balanza durante el tiempo suficiente para que los pesafiltros se enfríen totalmente, y en cualquier caso, durante un mínimo de 2 horas.
1.7.1.3. Pesado.
Después del enfriamiento, pesar los pesafiltros en los dos minutos siguientes al acto de sacarlos del desecador. Pesar con 0,0002 g de precisión.
I.7.2. Modo de operar.
Tomar de la muestra sometida a tratamiento previo una muestra de análisis con un peso mínimo de 1 g. Cortar el hilado o el tejido en partes de 10 mm de largo aproximadamente y disgregarlas lo mejor posible. Secar la muestra en un vidrio de reloj, enfriar en un desecador y pesar. Trasferir la muestra al recipiente de cristal indicado en la parte correspondiente del método comunitario; inmediatamente después, pesar de nuevo el pesafiltros y calcular por diferencia el peso en seco de la muestra. Completar el análisis del modo indicado en la parte correspondiente del método aplicable. Examinar el residuo al microscopio para cerciorarse de que el tratamiento haya eliminado completamente la fibra soluble.
I.8. Cálculo y presentación de los resultados.
Expresar el peso del componente insoluble en forma de porcentaje del peso total de las fibras presentes en la mezcla. El porcentaje del componente soluble se obtendrá por diferencia. Calcular los resultados sobre la base del peso en seco de las distintas fibras puras, ajustado con los porcentajes convencionales y con los factores de corrección necesarios para tener en cuenta las pérdidas de materia durante el tratamiento previo y el análisis.
Estos cálculos se harán aplicando la fórmula dada en el punto I.8.2.
I.8.1. Cálculo del porcentaje del peso del componente insoluble puro, no teniendo en cuenta la pérdida de peso sufrida por las fibras durante el tratamiento previo:
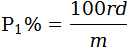
P1 es el porcentaje del componente insoluble seco y puro,
m es el peso en seco de la muestra después del tratamiento previo,
r es el peso en seco del residuo,
d es el factor de corrección que tiene en cuenta la pérdida de peso del componente insoluble en el reactivo durante el análisis. Los valores apropiados de «d» se dan en la parte correspondiente del texto de cada método
Estos valores de «d» son, por supuesto, los valores normales aplicables a las fibras no degradadas químicamente.
I.8.2. Cálculo del porcentaje del peso del componente insoluble, ajustado con los porcentajes convencionales y en su caso, con los factores de corrección de la pérdida de peso ocasionada por el tratamiento previo:
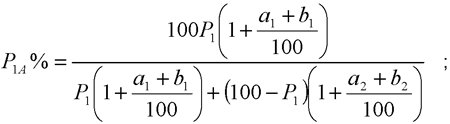
P1A es el porcentaje del componente insoluble teniendo en cuenta el porcentaje convencional y la pérdida de peso experimentada durante el tratamiento previo,
P1 es el porcentaje del componente insoluble seco y puro calculado con la fórmula indicada en el punto I.8.1,
a1 es el porcentaje convencional del componente insoluble (Anexo II del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.),
a2 es el porcentaje convencional del componente soluble (Anexo II del Real Decreto 928/1987, de 5 de junio, relativo al etiquetado de composición de los productos textiles.)
b1 es la pérdida porcentual del componente insoluble por efecto del tratamiento previo
b2 es la pérdida porcentual del componente soluble por efecto del tratamiento previo.
El porcentaje del segundo componente (P2A %) es igual a 100‑P1A %.
En caso de que se utilice un tratamiento previo especial, los valores de b1 y de b2 deberán determinarse, si fuera posible, sometiendo cada una de las fibras componentes puras al tratamiento previo aplicado durante el análisis. Se entiende por fibras puras, las fibras exentas de toda materia no fibrosa, a excepción de las que contengan normalmente (por su naturaleza o como consecuencia del proceso de fabricación) en el estado (crudo, blanqueado) en que se encuentren en el artículo sometido al análisis.
En caso de que no se disponga de fibras componentes separadas y puras que hayan servido para la fabricación del artículo sometido a análisis, se adoptarán los valores medios de b1 y de b2, resultantes de pruebas efectuadas en fibras puras parecidas a las que contenga la mezcla examinada.
Si se aplica el tratamiento previo normal por extracción con éter de petróleo y agua, podrán despreciarse los factores de corrección b1 y b2, salvo en los casos del algodón crudo, del lino crudo y del cáñamo crudo, en los que se admite convencionalmente que la pérdida debida al tratamiento previo es igual al 4 %, y salvo en el caso del polipropileno, en el que se admite convencionalmente que dicha pérdida es igual al 1 %.
En el caso de otras fibras, se admite convencionalmente que no se tengan en cuenta en los cálculos las pérdidas por efecto del tratamiento previo.
II.1. Ámbito de aplicación.
El método se aplicará a las fibras textiles, cualquiera que sea su naturaleza, a condición que no formen una mezcla íntima y que sea posible separarlas a mano.
II.2. Principio.
Después de haber identificado los componentes del tejido, se eliminarán primero las materias no fibrosas por un tratamiento previo apropiado y después se separarán las fibras a mano, se secarán y pesarán para calcular la proporción de cada fibra en la mezcla.
II.3. Material necesario.
II.3.1. pesafiltros o cualquier otro instrumental que dé idénticos resultados.
II.3.2. Desecador que contenga gel de sílice coloreado por medio de un indicador.
II.3.3. Horno de secado con ventilador para secar las muestras a 105 ºC ± 3 ºC.
II.3.4. Balanza analítica, sensibilidad 0,0002 g.
II.3.5. Equipo de extracción Soxhlet o instrumental que permita idéntico resultado.
II.3.6. Aguja.
II.3.7. Torsiómetro o aparato equivalente.
II.4. Reactivos.
II.4.1. Éter de petróleo redestilado con punto de ebullición entre 40 ºC y 60 ºC.
II.4.2. Agua destilada o desionizada.
II.5. Atmósfera de acondicionamiento y de análisis.
Ver el punto I.4.
II.6. Muestra reducida.
Ver el punto I.5.
II.7. Tratamiento previo de la muestra reducida.
Ver el punto I.6.
II.8. Procedimiento de análisis.
II.8.1. Análisis de un hilo.
Tomar de la muestra sometida a tratamiento previo una muestra de un peso mínimo de 1 g. En caso de un hilo muy fino, el análisis podrá efectuarse sobre un largo de 30 m como mínimo, cualquiera que sea su peso.
Cortar el hilo en trozos de longitud conveniente y separar las distintas fibras con la ayuda de una aguja y, si fuese necesario, del torsiómetro. Los tipos de fibras así separadas se pondrán en pesafiltros tarados y secados a 105 ºC ± 3 ºC hasta obtener una masa constante, tal y como se describe en los puntos I.7.1 y I.7.2.
II.8.2. Análisis de un tejido.
Tomar de la muestra sometida a tratamiento previo una muestra de un peso mínimo de 1 g, que no sea de la orilla, con bordes cortados con precisión, sin hilachas, y paralelos a los hilos de urdimbre o de trama o, en caso de tejidos de punto, paralelos a las hileras y a los hilos de los puntos. Separar los hilos de tipos diferentes, recogerlos en los pesafiltros tarados y proceder como se indica en el punto II.8.1.
II.9. Cálculo y presentación de los resultados.
Expresar el peso de cada uno de los componentes en forma de porcentaje del peso total de las fibras presentes en la mezcla. Calcular los resultados sobre la base del peso en seco de las distintas fibras puras, ajustado con los porcentajes convencionales y con los factores de corrección necesarios para tener en cuenta las pérdidas de masa por efecto del tratamiento previo.
II.9.1. Cálculo de los porcentajes del peso en seco de las distintas fibras depuradas, no teniendo en cuenta la pérdida de peso sufrida por las fibras por efecto del tratamiento previo:
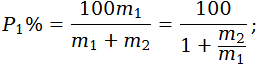
P1 es el porcentaje del primer componente seco y depurado,
m1 es la masa del primer componente seco y depurado,
m2 es la masa del segundo componente seco y depurado.
II.9.2. Cálculo de los porcentajes del peso en seco de cada uno de los componentes, ajustado con los porcentajes convencionales y en su caso, con los factores de corrección de las pérdidas de peso ocasionadas por el tratamiento previo (ver el punto I.8.2).
III.1. Precisión de los métodos.
La precisión indicada para cada método se relaciona con la reproductibilidad.
La reproducibilidad es la fidelidad, es decir, el mayor o menor grado de concordancia entre los valores experimentales obtenidos por operadores que trabajen en laboratorios diferentes o en épocas diferentes, y obtengan cada uno de ellos con el mismo método resultados individuales sobre un producto homogéneo idéntico.
La reproducibilidad se expresa por los márgenes de fiabilidad de los resultados, para un margen de fiabilidad del 95 %.
Se entiende con ello la diferencia entre dos resultados que, en una serie de análisis efectuados en diferentes laboratorios, sólo se superaría en el cinco por ciento de los casos, aplicando normal y correctamente el método a una mezcla homogénea idéntica.
III.2. Informe del análisis.
III.2.1. Indicar que el análisis se ha efectuado conforme al presente método.
III.2.2. Dar información detallada referente a los tratamientos previos especiales (ver el punto I.6).
III.2.3. Indicar los resultados individuales y la media aritmética con un decimal.
2. MÉTODOS PARTICULARES – CUADRO RESUMEN
|
Método |
Ámbito de aplicación |
Reactivo |
|
|---|---|---|---|
|
Nº 1 |
Acetato. |
Otras fibras determinadas. |
Acetona. |
|
Nº 2 |
Determinadas fibras proteínicas. |
Otras fibras determinadas. |
Hipoclorito. |
|
Nº 3 |
Viscosa, cupro o determinados tipos de modal. |
Algodón. |
Ácido fórmico y cloruro de cinc. |
|
Nº 4 |
Poliamida o nailon. |
Otras fibras determinadas. |
Ácido fórmico al 80%. |
|
Nº 5 |
Acetato. |
Triacetato. |
Alcohol bencílico. |
|
Nº 6 |
Triacetato o polilactida. |
Otras fibras determinadas. |
Diclorometano. |
|
Nº 7 |
Determinadas fibras celulósicas. |
Poliéster o elastomultiéster. |
Ácido sulfúrico al 75%. |
|
Nº 8 |
Acrílicos, determinados modacrílicos o determinadas clorofibras. |
Otras fibras determinadas. |
Dimetilformamida. |
|
Nº 9 |
Determinadas clorofibras. |
Otras fibras determinadas. |
Disulfuro de carbono/acetona, 55,5/44,5. |
|
Nº 10 |
Acetato. |
Determinadas clorofibras. |
Ácido acético glacial. |
|
Nº 11 |
Seda. |
Lana o pelo. |
Ácido sulfúrico al 75%. |
|
Nº 12 |
Yute. |
Determinadas fibras de origen animal. |
Determinación del contenido en nitrógeno. |
|
Nº 13 |
Polipropileno. |
Otras fibras determinadas. |
Xileno. |
|
Nº 14 |
Otras fibras determinadas. |
Clorofribras (a base de homo-polímero de cloruro de vinilo). |
Ácido sulfúrico concentrado. |
|
Nº 15 |
Clorofibras, determinados modacrílicos y elastanos, acetatos, triacetatos. |
Otras fibras determinadas. |
Ciclohexanona>. |
MÉTODO Nº 1
ACETATO Y OTRAS FIBRAS DETERMINADAS
(Método de la acetona)
1. ÁMBITO DE APLICACIÓN.
El presente método se aplicará, después de haber eliminado las materias no fibrosas, a mezclas binarias de:
1) acetato (19)
con
2) lana (1), pelo de animal (2 y 3), seda (4), algodón (5), lino (7), cáñamo (8), yute (9), abacá (10), esparto (11), coco (12), retama (13), ramio (14), sisal (15), cupro (21), modal (22), proteínica (23), viscosa (25), acrílico (26), poliamida o nailon (30), poliéster (34) y elastomultiéster (45).
Obviamente, este método no se aplicará al acetato desacetilizado en superficie.
2. PRINCIPIO.
Las fibras de acetato se disolverán con acetona a partir de una cantidad conocida de la mezcla en estado seco. Se recogerá el residuo, lavado, secado y pesado; su peso, corregido en su caso, se expresará en forma de porcentaje del peso en seco de la mezcla. El porcentaje de acetato en seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en el apartado generalidades).
3.1. Instrumental.
Frascos cónicos de una capacidad mínima de 200 ml, provistos de un tapón esmerilado.
3.2 Reactivo.
Acetona.
4. MODO DE OPERAR.
Aplicar el procedimiento descrito en las generalidades y proceder como sigue:
Añadir 100 ml de acetona por gramo de muestra contenida en el frasco cónico de una capacidad mínima de 200 ml provisto de un tapón esmerilado, agitar el frasco, dejar durante 30 minutos a temperatura ambiente agitando de vez en cuando y decantar después el líquido a través de la placa filtrante tarada.
Repetir este tratamiento dos veces más (en total tres extracciones), pero sólo durante 15 minutos cada vez, de manera que el tiempo total del tratamiento con acetona sea de una hora. Transvasar el residuo a la placa filtrante. Lavar el residuo en la placa filtrante por medio de acetona, ayudándose del vacío. Llenar de nuevo la placa filtrante de acetona que se dejará después filtrar naturalmente, sin succión.
Finalmente, escurrir la placa por medio del vacío, secar la placa y el residuo, enfriar y pesar.
5. CÁLCULO Y EXPRESIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 1,00.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con el método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 2
DETERMINADAS FIBRAS PROTEÍNICAS Y OTRAS FIBRAS
(Método del hipoclorito)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de haber eliminado las materias no fibrosas, a las mezclas binarias de:
1) determinadas fibras proteínicas, a saber: lana (1), pelo de animales (2 y 3), seda (4), proteínas (23)
con
2) algodón (5), cupro (21), viscosa (25), acrílico (26), clorofibras (27), poliamida o nailon (30), poliéster (34), polipropileno (36), elastano (42) fibra de vidrio (43) y elastomultiéster (45).
Si estuviesen presentes varias fibras proteínicas, el método permitirá determinar su cantidad total pero no su porcentaje individual.
2. PRINCIPIO.
Las fibras proteínicas se disolverán con una solución de hipoclorito a partir de una cantidad conocida de la mezcla en estado seco. Se recogerá el residuo, lavado, seco y pesado. Su peso, corregido si fuera necesario, se expresará en forma de porcentaje del peso en seco de la mezcla. El porcentaje de las fibras proteínicas secas se obtendrá por diferencia.
Para preparar la solución de hipoclorito puede utilizarse hipoclorito de litio o hipoclorito de sodio.
El hipoclorito de litio resulta indicado cuando el número de análisis es reducido o cuando los análisis se efectúan con largos intervalos de tiempo. El hipoclorito de litio sólido, contrariamente al hipoclorito de sodio, presenta un contenido en hipoclorito prácticamente constante. Si dicho contenido es conocido ya no resultará necesario determinarlo por iodometría en cada análisis y se podrá trabajar con tomas de ensayo de hipoclorito de litio constantes.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Frasco cónico de 250 ml con tapón de vidrio esmerilado.
ii) Termostato regulable a 20 ºC ± 2 ºC.
3.2. Reactivos.
i) Reactivos a base de hipoclorito.
a) Solución de hipoclorito de litio.
Este reactivo estará constituido por una solución recién preparada que contenga 35 (± 2) g/l de cloro activo (aproximadamente 1 M) a la que se habrá añadido hidróxido de sodio previamente disuelto a razón de 5 (± 0,5) g/l. Para preparar la solución disolver 100 g de hipoclorito de litio con un contenido en cloro activo del 35 % (o 115 g con un contenido en cloro activo del 30 %) en, aproximadamente, 700 ml de agua destilada. Añadir 5 g de hidróxido de sodio disuelto en, aproximadamente, 200 ml de agua destilada y completar hasta el litro con agua destilada. No es necesario controlar mediante iodometría esta solución recién preparada.
b) Solución de hipoclorito de sodio.
Este reactivo estará constituido por una solución recién preparada de un contenido en cloro activo de 35 (± 2) g/l (aproximadamente 1 M) a la que se habrá añadido hidróxido de sodio previamente disuelto a razón de 5 (± 0,5) g/l. Verificar mediante iodometría antes de cada análisis, la concentración en cloro activo de la solución.
ii) Ácido acético diluido.
Diluir 5 ml de ácido acético glacial en 1 l con agua.
4. MODO DE OPERAR.
Aplicar el procedimiento descrito en las generalidades y proceder como sigue:
Introducir aproximadamente 1 g de la muestra en el matraz de 250 ml; añadir alrededor de 100 ml de solución de hipoclorito (hipoclorito de litio o de sodio). Agitar enérgicamente para empapar bien la muestra.
A continuación colocar el matraz en un termostato a 20 ºC durante 40 minutos; durante dicho intervalo de tiempo agitar continuamente, o por los menos, frecuentemente y a intervalos regulares. Dado el carácter exotérmico de la disolución de la lana, el calor de reacción debe repartirse y evacuarse de esta manera a fin de evitar importantes errores provocados por el ataque de fibras no solubles.
Transcurridos los 40 minutos, filtrar el contenido del matraz mediante una placa filtrante tarada. Enjuagar el matraz con un poco de reactivo de hipoclorito a fin de eliminar las fibras que pudieran encontrarse aún en el mismo y verterlo todo en la placa filtrante. Vaciar la placa filtrante mediante vacío; lavar el residuo sucesivamente con agua, con el ácido acético diluido y nuevamente con agua. Durante dicha operación escurrir la placa filtrante mediante vacío después de cada adición de líquido esperando, sin embargo, que el líquido se haya filtrado por gravedad.
Finalmente escurrir la placa filtrante mediante vacío, secar la placa con el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados tal y como se describe en las generalidades. El coeficiente de corrección «d» tiene valor 1,00. Tiene valor 1,01 para el algodón, la viscosa y el modal, y valor 1,03 para el algodón crudo.
6. PRECISIÓN DEL MÉTODO.
En el caso de las mezclas homogéneas de fibras textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 3
VISCOSA, CUPRO O DETERMINADOS TIPOS DE MODAL Y ALGODÓN
(Método del ácido fórmico y del cloruro de zinc)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de haber eliminado las materias no fibrosas, a las mezclas binarias de:
1) viscosa (25) o cupro (21) incluidos determinados tipos de modal (22).
con
2) algodón (5).
Si se comprobase la presencia de una fibra modal, deberá efectuarse una prueba preliminar para comprobar si esta fibra es soluble en el reactivo.
Este método no se aplicará a las mezclas en las que el algodón haya sufrido una degradación química excesiva, ni cuando la viscosa o el cupro se hayan vuelto parcialmente insolubles por la presencia de colorantes o aprestos que no puedan eliminarse completamente.
2. PRINCIPIO.
Las fibras de viscosa, de cupro o de modal se disolverán, a partir de una cantidad conocida de la mezcla en estado seco, con un reactivo compuesto por ácido fórmico y cloruro de zinc. Se recogerá el residuo, lavado, secado y pesado; después de corrección, su peso se expresará en porcentaje del peso en seco de la mezcla. El porcentaje de viscosa, de cupro o de modal en seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los descritos en las generalidades)
3.1. Instrumental.
i) Frascos cónicos de una capacidad mínima de 200 ml provistos de tapón esmerilado.
ii) Dispositivo que permita mantener los frascos a 40 ºC ± 2 ºC.
3.2. Reactivos.
i) Solución que contenga 20 g de cloruro de zinc anhidro fundido y 68 g de ácido fórmico anhidro, llevada a 100 g con agua (es decir, 20 partes en peso de cloruro de zinc anhidro fundido en 80 partes en peso de ácido fórmico al 85 % en peso).
Nota:
A este respecto, se recuerda el punto I.3.2.2, que establece que todos los reactivos utilizados deben ser químicamente puros; además, debe utilizarse únicamente cloruro de zinc anhidro fundido.
ii) Solución de hidróxido de amonio:
Diluir 20 ml de una solución concentrada de amoniaco (0,880 g/l) en 980 ml de agua.
4. MODO DE OPERAR.
Aplicar el procedimiento descrito en las generalidades y proceder como sigue:
Introducir inmediatamente la muestra en el frasco, previamente calentado a 40 ºC. Añadir 100 ml de solución de ácido fórmico y de cloruro de zinc previamente calentada a 40 ºC, por g de muestra. Cerrar el frasco y agitar. Mantener el frasco y su contenido a 40 ºC durante dos horas y media agitando dos veces a intervalos de una hora. Filtrar el contenido del frasco a través de una placa filtrante tarada y con la ayuda del reactivo, transferir a ésta las fibras que pudieran haber quedado en el frasco. Enjuagar con 20 ml de reactivo.
Lavar a fondo la placa y el residuo con agua a 40 ºC. Hasta la total desaparición del amoniaco aclarar el residuo fibroso con 100 ml aproximadamente de solución de amoniaco fría (3.2.ii), asegurándose que este residuo permanezca totalmente sumergido en la solución durante 10 minutos8; aclarar después a fondo con agua fría.
8 Para asegurar que el residuo fibroso permanece sumergido, en la solución de amoniaco se puede, por ejemplo, adaptar a la placa filtrante un alargador con grifo que permita regular el paso del amoniaco.
No aplicar el vacío hasta que cada solución de lavado no se haya filtrado por gravedad. Eliminar finalmente el líquido restante por medio del vacío, secar la placa y su residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» para el algodón es de 1,02.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 2, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 4
POLIAMIDA O NAILON Y OTRAS FIBRAS DETERMINADAS
(Método del ácido fórmico al 80 %)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) poliamida o nailon (30)
con
2) lana (1), pelo de animal (2 y 3), algodón (5), cupro (21), modal (22), viscosa (25), acrílico (26), clorofibras (27), poliéster (34), polipropileno (36) fibra de vidrio (43) y elastomuliéster (45).
Como se acaba de indicar, este método se aplicará a las mezclas que contengan lana, pero cuando la proporción de esta última sea superior al 25 %, deberá aplicarse el método n° 2 (disolución de la lana en solución de hipoclorito de sodio alcalino).
2. PRINCIPIO.
Las fibras de poliamida se disolverán con ácido fórmico a partir de una cantidad conocida de la mezcla en estado seco. El residuo se recogerá, lavado, secado y pesado. Corregido su peso si fuese necesario, se expresará en porcentaje del peso en seco de la mezcla. El porcentaje de poliamida o nailon en seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los descritos en las generalidades)
3.1. Instrumental.
Frasco cónico de una capacidad mínima de 200 ml, provisto de un tapón esmerilado.
3.2. Reactivos.
i) Ácido fórmico al 80 % m/m (densidad a 20 ºC: 1,186):
Llevar 880 ml de ácido fórmico al 90 % m/m (densidad a 20 ºC: 1,204), a 1 l con agua, o bien llevar 780 ml de ácido fórmico al 98-100 % m/m (densidad a 20 ºC: 1,220), a 1 l con agua.
La concentración no es crítica entre 77 y 83 % m/m de ácido fórmico.
ii) Amoniaco diluido:
Llevar 80 ml de amoniaco concentrado (densidad a 20 ºC: 0,880), a 1 l con agua.
4. MODO DE OPERAR
Seguir el proceso descrito en las generalidades y proceder como sigue:
Añadir a la muestra, contenida en el frasco cónico de 200 ml de capacidad mínima, 100 ml de ácido fórmico por gramo de muestra. Tapar y agitar para empapar la muestra. Dejar reposar durante 15 minutos a temperatura ambiente, agitando de vez en cuando. Filtrar el contenido del frasco en una placa filtrante tarada y pasar a ésta las posibles fibras residuales lavando el frasco con un poco de ácido fórmico. Escurrir la placa por succión y lavar el residuo sobre la placa, sucesivamente con ácido fórmico, agua caliente, amoniaco diluido y por último con agua fría. Escurrir el crisol por succión después de cada adición. No aplicar el vacío hasta que cada solución de lavado no se haya filtrado por gravedad. Finalmente, escurrir el crisol por succión, secarlo con el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 1,00.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 5.
ACETATO Y TRIACETATO
(Método del alcohol bencílico)
1. ÁMBITO DE APLICACIÓN.
El método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
– acetato (19).
con
– triacetato (24).
2. PRINCIPIO.
Las fibras de acetato se disolverán, a partir de una cantidad conocida de la mezcla en estado seco, con alcohol bencílico a 52 ºC ± 2 ºC.
Se recogerá el residuo, lavado, secado y pesado; se expresará su peso en porcentaje del peso en seco de la mezcla. El porcentaje de acetato en seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Frasco cónico de una capacidad mínima de 200 ml provisto de un tapón esmerilado.
ii) Agitador mecánico.
iii) Termostato o cualquier otro aparato que permita mantener el frasco a la temperatura de 52 ºC ± 2 ºC.
3.2. Reactivos.
i) Alcohol bencílico.
ii) Alcohol etílico.
4. MODO DE OPERAR.
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en el frasco cónico 100 ml de alcohol bencílico por gramo de muestra.
Poner el tapón, fijar el frasco al agitador de tal manera que se sumerja en un baño de agua mantenido a 52 ºC ± 2 ºC y agitar durante 20 minutos a esta temperatura (se podrá ocasionalmente sustituir la agitación mecánica por una enérgica agitación manual).
Decantar el líquido a través de la placa filtrante tarada. Añadir al frasco una nueva dosis de alcohol bencílico y agitar de nuevo a 52 ºC ± 2 ºC durante veinte minutos.
Decantar a través de la placa. Repetir este ciclo de operaciones una tercera vez.
Finalmente, verter el líquido y el residuo en la placa y transferir a ésta las fibras que pudieran quedar en el frasco, utilizando para ello una cantidad suplementaria de alcohol bencílico a 52 ºC ± 2 ºC.
Escurrir bien la placa. Transferir las fibras a un frasco, enjuagar con alcohol etílico y después de agitar manualmente, decantar a través de la placa filtrante.
Repetir esta operación de enjuague dos o tres veces. Transferir el residuo a la placa y escurrirla a fondo. Secar la placa y el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 1,00.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 6
TRIACETATO Y OTRAS FIBRAS DETERMINADAS
(Método del diclorometano)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) triacetato (24) o polilactida (33a)
con
2) lana (1), pelo animal (2 y 3), seda (4), algodón (5), cupro (21), modal (22), viscosa (25), acrílico (26), poliamida o nailon (30), poliéster (34), fibra de vidrio (43) y elastomultiéster (45).
Nota:
Las fibras de triacetato parcialmente saponificadas por un apresto especial dejan de ser completamente solubles en el reactivo. En este caso, el método no será aplicable.
2. PRINCIPIO.
Las fibras de triacetato o polilactida con diclorometano se disolverán a partir de una cantidad conocida de la mezcla en estado seco. El residuo se recogerá, se lavará, se secará y se pesará; su masa, corregida si fuese necesario, se expresará en porcentaje de la masa de la mezcla en estado seco. El porcentaje de triacetato o polilactida en seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
Frasco cónico de una capacidad mínima de 200 ml provisto de un tapón esmerilado.
3.2. Reactivo.
Diclorometano (cloruro de metileno).
4. MODO DE OPERAR.
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en un frasco cónico de 200 ml provisto de un tapón esmerilado, 100 ml de diclorometano por gramo de muestra; poner el tapón, agitar el frasco cada diez minutos para empapar bien la muestra y dejar reposar el frasco durante 30 minutos a temperatura ambiente, agitando a intervalos regulares. Decantar el líquido a través de la placa filtrante tarada. Añadir 60 ml de diclorometano al frasco que contenga el residuo, agitar manualmente y filtrar el contenido del frasco a través de la placa filtrante. Transferir a ésta las fibras residuales con ayuda de una pequeña cantidad suplementaria de diclorometano. Aplicar el vacío a la placa para eliminar el exceso de líquido, llenarla de nuevo de diclorometano y dejar que éste se filtre por gravedad.
Finalmente, aplicar el vacío para eliminar el exceso de líquido, tratar a continuación el residuo con agua hirviendo para eliminar todo el disolvente, aplicar el vacío, secar la placa y el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados según lo descrito en las generalidades. El valor de «d» es de 1,00, salvo para el poliéster y el elastomultiéster, para los que el valor de «d» es de 1,01.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 7
DETERMINADAS FIBRAS CELULÓSICAS Y POLIÉSTER
(Método del ácido sulfúrico al 75 %).
1. ÁMBITO DE APLICACIÓN.
El método se aplicará, después de la eliminación de materias no fibrosas, a las mezclas binarias de:
1) algodón (5), lino (7), cáñamo (8), ramio (14), cupro (21), modal (22), viscosa (25)
con
2) poliéster (34) y elastomultiéster (45).
2. PRINCIPIO.
Las fibras celulósicas se disolverán con ácido sulfúrico al 75 % a partir de una cantidad conocida de la mezcla en estado seco. El residuo se recogerá, lavado, secado y pesado; su peso se expresará en porcentaje del peso de la mezcla en estado seco. La proporción de fibras celulósicas secas se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Frasco cónico de una capacidad mínima de 500 ml, provisto de un tapón esmerilado.
ii) Termostato o cualquier otro aparato que permita mantener el frasco a temperatura de 50 ºC ± 5 ºC.
3.2. Reactivos.
i) Ácido sulfúrico al 75 % ± 2 % m/m:
Añadir con cuidado y enfriándolo, 700 ml de ácido sulfúrico de densidad 1,84 a 20 ºC, a 350 ml de agua destilada. Una vez enfriada la solución a temperatura ambiente, llevar el volumen a 1 1 con agua.
ii) Solución de amoniaco diluida:
Diluir 80 ml de solución de amoniaco de densidad 0,88 a 20 ºC en 920 ml de agua.
4. MODO DE OPERAR
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en el frasco cónico de 500 ml de capacidad mínima provisto de un tapón esmerilado, 200 ml de ácido sulfúrico al 75 % por gramo de muestra; poner el tapón y agitar con cuidado el frasco para empapar bien la toma de prueba. Mantener el frasco a 50 ºC ± 5 ºC durante una hora agitando a intervalos regulares de aproximadamente 10 minutos. Filtrar por succión el contenido del frasco a través de una placa filtrante tarada. Transferir a ésta las fibras residuales enjuagando el frasco con un poco de ácido sulfúrico al 75 %. Escurrir la placa mediante succión y lavar una vez el residuo que se encuentre en la placa, llenando ésta de ácido sulfúrico al 75 %. No aplicar el vacío hasta que el ácido no se haya filtrado por gravedad.
Lavar el residuo varias veces con agua fría, dos veces con la solución de amoníaco diluida, y después a fondo con agua fría, escurriendo la placa por succión después de cada adición. No aplicar el vacío hasta que cada una de las soluciones de lavado no se haya filtrado por gravedad. Eliminar en fin el líquido restante por medio del vacío, secar la placa y el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 1,00.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores al ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 8
ACRÍLICAS, DETERMINADAS MODACRÍLICAS O DETERMINADAS CLOROFIBRAS Y OTRAS FIBRAS DETERMINADAS
(Método del dimetilformamida)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) acrílicos (26), determinados modacrílicos (29) o determinadas clorofibras (27)9
9 Antes de proceder al análisis se deberá comprobar la solubilidad de esas modacrílicas o clorofibras en el reactivo.
con
2) lana (1), pelo animal (2 y 3), seda (4), algodón (5), cupro (21), modal (22), viscosa (25), poliamida o nailon (30), poliéster (34) y elastomultiéster (45).
Se aplicará igualmente a los acrílicos y a determinados modacrílicos tratados con colorantes premetalizados, pero no a los tratados con colorantes cromotrópicos.
2. PRINCIPIO.
Las fibras acrílicas, determinadas modacrílicas o determinadas clorofibras se disolverán a partir de una cantidad conocida de la mezcla en estado seco por medio de dimetilformamida a temperatura de baño María hirviendo. El residuo se recogerá, lavado, secado y pesado. Su peso, corregido si fuese necesario, se expresará en porcentaje del peso de la mezcla en estado seco y el porcentaje de acrílicos, modacrílicos o de clorofibras secas se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Frasco cónico de una capacidad mínima de 200 ml, provisto de un tapón esmerilado.
ii) Baño María en ebullición.
3.2. Reactivo.
Dimetilformamida (punto de ebullición 153 ºC ± 1 ºC) que no contenga más de un 0,1 % de agua.
Se recomienda trabajar con una protección adecuada, ya que este reactivo es tóxico.
4. MODO DE OPERAR.
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en el frasco cónico 80 ml de dimetilformamida por gramo de muestra calentada previamente al baño María hirviendo, tapar, agitar para que la toma de prueba se empape bien y mantener en el baño María hirviendo durante una hora.
Agitar manualmente el frasco y su contenido cinco veces durante este tiempo, procediendo con cuidado.
Decantar el líquido a través de una placa filtrante tarada, manteniendo las fibras en el frasco cónico. Añadir al frasco 60 ml de dimetilformamida y calentar de nuevo durante 30 minutos. Durante este tiempo, agitar manualmente el frasco y su contenido dos veces, procediendo con cuidado.
Filtrar por medio del vacío el contenido del frasco a través de la placa filtrante.
Transferir las fibras residuales a la placa filtrante enjuagando el matraz con dimetilformamida. Aplicar el vacío para eliminar el exceso de líquido. Lavar el residuo con aproximadamente 1 l de agua caliente a 70-80 ºC. La placa filtrante debe llenarse de agua cada vez. Después de cada adición de agua aplicar brevemente el vacío, pero sólo después de que el agua se haya filtrado por gravedad. Si el líquido de lavado filtra demasiado lentamente a través de la placa filtrante se podrá aplicar el vacío ligeramente.
Secar la placa filtrante con el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados según lo descrito en las generalidades. El valor de «d» es de 1,00, salvo en los casos siguientes:
Lana 1,01
Algodón 1,01
Cupro 1,01
Modal 1,01
Poliéster 1,01
Elastomultiéster 1,01
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 9
DETERMINADAS CLOROFIBRAS Y OTRAS FIBRAS DETERMINADAS
(Método del sulfuro de carbono/acetona 55,5/44,5)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) determinadas clorofibras (27), a saber, determinados policloruros de vinilo, superclorados o no10
10 Antes de proceder al análisis se deberá comprobar la solubilidad de esas modacrílicas o clorofibras en el reactivo.
con
2) lana (1), pelo animal (2 y 3), seda (4), algodón (5), cupro (21), modal (22), viscosa (25), acrílico (26), poliamida o nailon (30), poliester (34), fibra de vidrio (43) y elastomultiéster (45).
Si el contenido en lana o en seda de la mezcla sobrepasase el 25 %, convendrá utilizar el método n° 2.
Si el contenido en poliamida o nailon sobrepasase el 25 % en la mezcla, se utilizará el método n° 4.
2. PRINCIPIO.
Se disolverán las fibras de clorofibras a partir de una cantidad conocida de la mezcla en estado seco con ayuda de la mezcla azeotrópica de sulfuro de carbono y de acetona. El residuo se recogerá, lavado, secado y pesado; su peso, corregido si fuese necesario, se expresará en porcentaje del peso de la mezcla en estado seco. El porcentaje de fibras de policloruro de vinilo secas se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los descritos en las generalidades).
3.1. Instrumental.
i) Frasco cónico de una capacidad mínima de 200 ml provisto de un tapón esmerilado.
ii) Agitador mecánico.
3.2. Reactivos.
i) Mezcla azeotrópica de sulfuro de carbono y de acetona (55,5 % de sulfuro de carbono y 44,5 % de acetona en volumen). Se recomienda trabajar con una protección adecuada ya que este reactivo es tóxico.
ii) Alcohol etílico al 92 % en volumen, o alcohol metílico.
4. MODO DE OPERAR.
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en el frasco cónico de 200 ml de capacidad mínima provisto de un tapón esmerilado, 100 ml de mezcla azeotrópica por gramo de muestra. Tapar convenientemente el frasco y agitar a temperatura ambiente durante 20 minutos por medio del agitador mecánico, o manualmente de manera vigorosa. Decantar el líquido flotante a través de la placa filtrante tarada.
Repetir el tratamiento con otros 100 ml de disolvente. Continuar este ciclo de operaciones hasta que una gota de líquido de extracción colocada sobre un vidrio de reloj no deje depósito de polímero después de evaporarse. Transferir el residuo a una placa filtrante con ayuda de una cantidad suplementaria de disolvente, aplicar el vacío para eliminar el líquido y enjuagar el crisol y el residuo, primero con 20 ml de alcohol y después, tres veces con agua. Dejar que la solución de lavado se filtre por gravedad antes de aplicar el vacío para eliminar el resto del líquido. Secar el crisol y el residuo, enfriar y pesar.
Nota:
Las muestras de ciertas mezclas con alto contenido de policloruro de vinilo encogen notablemente durante la operación de secado, lo que conlleva un retraso en la disolución del policloruro de vinilo por el disolvente. Sin embargo, esta contracción no impide la disolución total del policloruro de vinilo.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 1,00.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 10
ACETATO Y DETERMINADAS CLOROFIBRAS
(Método del ácido acético glacial)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) acetato (19).
con
2) algunas clorofibras (27), a saber, el policloruro de vinilo superclorado o no.
2. PRINCIPIO.
Las fibras de acetato se disolverán a partir de una cantidad conocida de la mezcla en estado seco con ácido acético glacial. El residuo se recogerá, lavado, secado y pesado; su peso, corregido cuando sea necesario, se expresará en porcentaje del peso de la mezcla en estado seco. El porcentaje de acetato seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los descritos en las generalidades).
3.1. Instrumental.
i) Frasco cónico de una capacidad mínima de 200 ml, provisto de un tapón esmerilado.
ii) Agitador mecánico.
3.2. Reactivo.
Ácido acético glacial (más del 99 %). Este reactivo deberá manipularse con precaución, ya que es extremadamente cáustico.
4. MODO DE OPERAR.
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en un frasco cónico de 200 ml de capacidad mínima provisto de un tapón esmerilado, 100 ml de ácido acético glacial por gramo de muestra. Tapar convenientemente el frasco y agitar durante 20 minutos a temperatura ambiente por medio del agitador mecánico, o manualmente de manera vigorosa. Decantar el líquido de la superficie a través de la placa filtrante tarada. Repetir este tratamiento dos veces más utilizando cada vez 100 ml de disolvente, de modo que se efectúen tres extracciones en total. Transferir el residuo a la placa filtrante, aplicar el vacío para eliminar el líquido y enjuagar la placa y el residuo, primero con 50 ml de ácido acético glacial, y después, tres veces con agua. Después de cada enjuagado dejar que la solución se filtre por gravedad antes de aplicar el vacío. Secar la placa y el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular el resultado de la manera descrita en las generalidades. El valor de «d» es de 1,00.
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 11
SEDA Y LANA O PELOS
(Método del ácido sulfúrico al 75 %)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) seda (4).
con
2) lana (1) o pelos de animales (2 y 3).
2. PRINCIPIO.
Las fibras de seda se disolverán a partir de una cantidad conocida de la mezcla en estado seco por medio de ácido sulfúrico al 75 % m/m11.
11 Las sedas silvestres, como el tusor, no son totalmente solubles en ácido sulfúrico al 75 %.
El residuo se recogerá, lavado, secado y pesado; su peso, corregido si fuese necesario, se expresará en porcentaje del peso total de la mezcla en estado seco. El porcentaje de seda en seco se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los descritos en las generalidades)
3.1. Instrumental.
Frascos cónicos de una capacidad mínima de 200 ml provistos de un tapón esmerilado.
3.2. Reactivos.
i) Ácido sulfúrico al 75 % ± 2 % m/m:
Añadir con cuidado y enfriándolo, 700 ml de ácido sulfúrico de densidad 1,84 a 20 ºC, a 350 ml de agua destilada. Después de enfriar hasta alcanzar la temperatura ambiente, llevar el volumen a un litro con agua.
ii) Ácido sulfúrico diluido: añadir lentamente 100 ml de ácido sulfúrico, de densidad 1,84 a 20 ºC, a 1 900 ml de agua destilada.
iii) Amoniaco diluido: llevar 200 ml de amoniaco concentrado, de densidad 0,880 a 20 ºC, a 1 000 ml con agua.
4. MODO DE OPERAR.
Seguir las instrucciones dadas en las generalidades y proceder como sigue:
Añadir a la toma de prueba contenida en un frasco cónico de 200 ml como mínimo provisto de un tapón esmerilado, 100 ml de ácido sulfúrico al 75 % por gramo de muestra, y tapar. Agitar vigorosamente y dejar 30 minutos a temperatura ambiente. Agitar de nuevo y dejar otros 30 minutos. Agitar una última vez y filtrar el contenido del frasco a través de la placa filtrante tarada. Arrastrar las fibras que pudieran quedar en la placa con ácido sulfúrico al 75 %. Lavar el residuo sobre el crisol, sucesivamente con 50 ml de ácido sulfúrico diluido, 50 ml de agua, y 50 ml de amoniaco diluido. Dejar cada vez las fibras en contacto con el líquido durante aproximadamente 10 minutos antes de aplicar el vacío. Aclarar finalmente con agua dejando las fibras en contacto con el agua durante 30 minutos aproximadamente. Aplicar el vacío para eliminar el resto del líquido. Secar el crisol y el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 0,985 para la lana (7).
6. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1 para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 12
YUTE Y ALGUNAS FIBRAS DE ORIGEN ANIMAL
(Método por determinación del contenido en nitrógeno)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) yute (9).
con
2) algunas fibras de origen animal.
Estas últimas podrán estar constituidas por pelos (2 y 3) o por lana (1) o por una mezcla de pelos y de lana. Este método no se aplicará a mezclas textiles que lleven materias no fibrosas (colorantes, aprestos, etc.) a base de nitrógeno.
2. PRINCIPIO.
Se determinará el contenido en nitrógeno de la mezcla, y se calculará, a partir de este dato y del contenido en nitrógeno conocido de los dos componentes, la proporción de cada uno de los componentes de la mezcla.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Matraz de digestión Kjeldahl de 200 a 300 ml.
ii) Aparatos de destilación Kjeldahl por arrastre de vapor.
iii) Instrumental de valoración por retroceso que permita una precisión de 0,05 ml.
3.2. Reactivos.
i) Tolueno.
ii) Metanol.
iii) Ácido sulfúrico de densidad 1,84 a 20 ºC12.
12 Estos reactivos carecerán de nitrógeno.
iv) Sulfato de potasio (9).
v) Bióxido de selenio (10).
vi) Solución de hidróxido de sodio (400 g/litro). Disolver 400 g de hidróxido de sodio en 400- 500 ml de agua y llevar a un litro con agua.
vii) Mezcla de indicadores:
Disolver 0,1 g de rojo de metilo en 95 ml de etanol y 5 ml de agua, y mezclar esta solución con 0,5 g de verde de bromocresol disuelto en 475 ml de etanol y 25 ml de agua.
viii) Solución de ácido bórico:
Disolver 20 g de ácido bórico en un litro de agua.
ix) Ácido sulfúrico 0,02 N (solución volumétrica patrón).
4. TRATAMIENTO PREVIO DE LA MUESTRA REDUCIDA.
El tratamiento previo descrito en las generalidades se sustituirá por el tratamiento previo siguiente:
En un aparato Soxhlet tratar la muestra secada al aire con una mezcla de un volumen de tolueno y de tres volúmenes de metanol durante cuatro horas, a una cadencia mínima de cinco ciclos por hora. Exponer la mezcla al aire para permitir la evaporación del disolvente y eliminar los últimos restos de éste por calentamiento en un horno a 105 ºC ± 3 ºC. Tratar después la muestra en agua (50 ml/g de muestra), haciéndola hervir por reflujo durante 30 minutos. Filtrar, reintroducir la muestra en el frasco y repetir la extracción con un volumen idéntico de agua. Filtrar, eliminar el exceso de agua de la muestra, por estrujamiento, succión o centrifugación y secar después la muestra al aire.
Nota:
El tolueno y el metanol son tóxicos. Conviene utilizar estos productos con gran prudencia.
5. MODO DE OPERAR.
5.1. Instrucciones generales.
En lo que se refiere a la toma, el secado y la pesada de la muestra, seguir el procedimiento descrito en las generalidades.
5.2. Procedimientos detallados.
Transferir al matraz de digestión Kjeldahl, una muestra que pese como mínimo 1 g. Añadir a la muestra contenida en el matraz de digestión y respetando el orden siguiente, 2,5 g de sulfato de potasio, 0,1-0,2 g de bióxido de selenio y 10 ml de ácido sulfúrico (d = 1,84). Calentar el matraz, primero lentamente, hasta la destrucción total de las fibras, después a fuego más fuerte, hasta que la solución se vuelva clara y prácticamente incolora. Continuar calentando durante 15 minutos. Dejar enfriar el matraz, añadir con cuidado al contenido 10-20 ml de agua, enfriar, transferir el contenido cuantitativamente a un matraz aforado de 200 ml y enrasar con agua para obtener la solución de análisis.
Introducir alrededor de 20 ml de solución de ácido bórico en un frasco cónico de 100 ml y situar este último bajo el refrigerador del aparato de destilación Kjeldahl de manera que el tubo de salida quede sumergido justo por debajo de la superficie de la solución de ácido bórico. Transferir exactamente 10 ml de la solución de análisis al matraz de destilación, introducir un mínimo de 5 ml de solución de hidróxido de sodio en el embudo, levantar ligeramente el tapón y dejar que la solución de hidróxido de sodio caiga lentamente en el matraz. Si la solución de análisis y la solución de hidróxido de sodio tendiesen a formar dos capas diferentes, mezclarlas agitando con cuidado. Calentar ligeramente el matraz de destilación e introducir en el líquido el vapor procedente del generador. Recoger 20 ml aproximadamente del destilado, bajar el frasco cónico de manera que la extremidad del tubo del refrigerador quede situada a unos 20 mm por debajo de la superficie del líquido y destilar durante un minuto más. Aclarar la extremidad del tubo con agua, recogiendo el líquido de lavado en el frasco cónico. Retirar este último y colocar un segundo frasco cónico que contenga alrededor de 10 ml de solución de ácido bórico y a continuación recoger aproximadamente 10 ml del destilado.
Valorar separadamente ambos destilados con ácido sulfúrico 0,02 N utilizando la mezcla de indicadores. Anotar los resultados de las respectivas valoraciones. Si la valoración del segundo destilado diese un resultado superior a 0,2 ml, repetir la prueba y comenzar de nuevo la destilación utilizando otra parte alícuota de la solución de análisis.
Efectuar una prueba en blanco, sometiendo a la digestión y a la destilación únicamente los reactivos.
6. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
6.1. Calcular el porcentaje de nitrógeno contenido en la muestra en estado seco del modo siguiente:
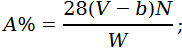
A % = porcentaje de nitrógeno en la muestra seca y pura,
V = volumen total en ml de ácido sulfúrico patrón utilizado para la determinación,
b = volumen total en ml de ácido sulfúrico patrón utilizado para la prueba en blanco,
N = valoración real del ácido sulfúrico patrón,
W = peso (g) de la toma de prueba en estado seco.
6.2. Aplicando valores del 0,22 % para el contenido en nitrógeno del yute, y del 16,2 % para el de las fibras de origen animal (ambos porcentajes expresados sobre el peso en seco de las fibras), calcular la composición de la mezcla con ayuda de la siguiente fórmula:
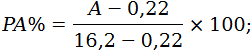
PA % = porcentaje de fibras de origen animal en la muestra.
7. PRECISIÓN DEL MÉTODO.
Sobre mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con este método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 13
POLIPROPILENO Y OTRAS FIBRAS DETERMINADAS
(Método al xileno)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, una vez eliminadas las materias no fibrosas, a las mezclas binarias de fibras de:
1) polipropileno (36).
con
2) lana (1), pelo animal (2 y 3), seda (4), algodón (5), acetato (19), cupro (21), modal (22), triacetato (24), viscosa (25), acrílico (26), poliamida o nailon (30), poliéster (34), fibra de vidrio (43) y elastomultiéster (45).
2. PRINCIPIO.
La fibra de polipropileno se disolverá a partir de una masa conocida de la mezcla en estado seco por disolución en xileno en ebullición. El residuo se recogerá, lavado, secado y pesado; su peso, corregido si fuese necesario, se expresará en porcentaje de la masa de la mezcla en estado seco. El porcentaje de polipropileno se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Frascos cónicos, capacidad mínima 200 ml, provistos de un tapón esmerilado;
ii) Refrigerante de reflujo (adaptado a líquidos de punto de ebullición elevado) con esmerilado adaptable a los frascos cónicos i).
3.2. Reactivo.
Xileno que destile entre 137 ºC y 142 ºC.
Nota:
Dicho reactivo es muy inflamable y produce vapores tóxicos. Deberán tomarse precauciones cuando se utilice.
4. MODO DE OPERAR.
Seguir el procedimiento descrito en las generalidades, y proceder después de la forma siguiente:
A la toma de muestra colocada en el frasco cónico [3.1.i)] añadir 100 ml de xileno (3.2) por gramo de toma de muestra. Colocar el refrigerante [3.1.ii)] y llevar a ebullición, que se mantendrá durante 3 minutos. Decantar inmediatamente el líquido caliente en la placa filtrante tarada (véase la nota 1). Repetir dicho tratamiento dos veces más utilizando cada vez 50 ml de disolvente.
Lavar el residuo que quede en el frasco con 30 ml de xileno hirviendo (dos veces), y luego, también dos veces, con 75 ml cada vez de éter de petróleo (I.3.2.1 de las generalidades).
Tras el segundo lavado con éter de petróleo, filtrar el contenido del frasco a través de la placa filtrante y transferir las fibras residuales a la placa con ayuda de una pequeña cantidad suplementaria de éter de petróleo. Hacer que se evapore completamente el disolvente. Secar la placa y el residuo, enfriarlos y pesarlos.
Notas:
1) La placa filtrante en la que se decante el xileno deberá calentarse previamente.
2) Después del tratamiento con xileno hirviendo, cerciorarse de que el frasco que contenga el residuo está lo suficientemente frío antes de introducir en él el éter de petróleo.
3) Para evitar a los analistas los peligros derivados de la inflamabilidad y toxicidad de los productos que se manejan podrán utilizarse aparatos de extracción al calor y modos de operar apropiados que den resultados idénticos13.
13 Véase, por ejemplo, el material descrito en Melliand Textilberichte 56 (1975) pp. 643-645.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la forma descrita en las generalidades. El valor de «d» es 1,00.
6. PRECISIÓN DEL MÉTODO.
Con una mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con dicho método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 14
CLOROFIBRAS (A BASE DE HOMOPOLÍMERO DE CLORURO DE VINILO) Y OTRAS FIBRAS DETERMINADAS
(Método al ácido sulfúrico concentrado)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, una vez eliminadas las materias no fibrosas, a las mezclas binarias de:
1) clorofibras (27) a base de homopolímero de cloruro de vinilo (sobredorado o no)
con
2) algodón (5), acetato (19), cupro (21), modal (22), triacetato (24), viscosa (25), determinados acrílicos (26), determinados modacrílicos (29), poliamida o nailon (30), poliéster (34) y elastomultiéster (45).
Los modacrílicos de que se trata son los que dan una solución límpida por inmersión en ácido sulfúrico concentrado (d20 = 1,84 g/ml).
Dicho método podrá utilizarse concretamente en sustitución de los métodos n° 8 y n° 9.
2. PRINCIPIO.
Las fibras mencionadas en el número 2 del apartado se eliminarán a partir de una masa conocida de la mezcla en estado seco por disolución en ácido sulfúrico concentrado d20 = 1,84 g/ml). El residuo, constituido por la clorofibra, se recogerá, lavado, secado y pesado; su peso, corregido si fuera necesario, se expresará en porcentaje del peso de la mezcla en estado seco. La proporción del segundo constituyente se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Frascos cónicos, capacidad mínima 200 ml, provistos de un tapón esmerilado.
ii) Varilla de vidrio de extremo plano.
3.2. Reactivos.
i) Ácido sulfúrico concentrado (d20 = 1,84 g/ml).
ii) Ácido sulfúrico, solución acuosa, alrededor de 50 % (m/m) de ácido sulfúrico.
Para preparar este reactivo, añadir, con precaución y enfriándolo 400 ml de ácido sulfúrico d20= 1,84 g/ml) a 500 ml de agua. Cuando se haya enfriado la solución a temperatura ambiente, llevar hasta un litro con agua.
iii) Amoniaco, solución diluida.
Diluir con agua destilada 60 ml de una solución de amoniaco concentrado (d20= 0,880 g/ml) para obtener un litro.
4. MODO DE OPERAR.
Seguir el procedimiento descrito en las generalidades, y proceder después de la forma siguiente:
A la toma de muestra colocada en el frasco [3.1.i)] añadir 100 ml de ácido sulfúrico [3.2.i)] por gramo de toma de muestra.
Dejar diez minutos a temperatura ambiente; agitando de vez en cuando la toma de muestra con ayuda de la varilla de vidrio. Si se tratara de una tela o de un tejido de punto, aplastarlo contra la pared del frasco y ejercer una ligera presión con ayuda de la varilla de vidrio para que la materia disuelta se separe con el ácido sulfúrico.
Decantar el líquido en la placa filtrante tarada. Añadir al frasco otros 100 ml de ácido sulfúrico [3.2.i)] y repetir la misma operación. Verter el contenido del frasco en la placa y transferir a ella el residuo fibroso con ayuda de la varilla de vidrio. Si fuera necesario, añadir al frasco un poco de ácido sulfúrico concentrado [3.2.i)] para arrastrar las fibras que hubieran podido quedar adheridas a las paredes. Vaciar la placa por aspiración; vaciar el filtrado del frasco o cambiar de frasco, lavar luego el residuo en la placa primero con la solución de ácido sulfúrico a 50 % [3.2.ii)], después con agua destilada o desionizada (I.3.2.3 de las generalidades), a continuación con la solución de amoniaco [3.2.iii)], y por último lavar a fondo con agua destilada o desionizada, vaciando completamente la placa mediante aspiración después de cada adición (no aplicar la aspiración durante la operación de lavado, esperar a que el líquido haya escurrido por gravedad).
Secar la placa y el residuo, enfriarlos y pesarlos.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la forma indicada en las generalidades. El valor de «d» es 1,00.
6. PRECISIÓN DEL MÉTODO.
Con una mezcla homogénea de materias textiles, los márgenes de fiabilidad de los resultados obtenidos con dicho método no serán superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
MÉTODO Nº 15
CLOROFIBRA, DETERMINADOS MODACRÍLICOS Y ELASTANOS, ACETATO, TRIACETATO Y OTRAS FIBRAS
(Método de la ciclohexanona)
1. ÁMBITO DE APLICACIÓN.
Este método se aplicará, después de la eliminación de las materias no fibrosas, a las mezclas binarias de:
1) acetato (19), triacetato (24), clorofibras (27), determinados modacrílicos (29) y determinados elastanos (39)
con
2) lana (1), pelos de animales (2 y 3), seda (4), algodón (5), cupro (21), modal (22), viscosa (25), poliamida o nailon (30), acrílico (26) y vidrio textil (40).
Si se observa la presencia de una fibra modacrílica o elastana, se deberá proceder a efectuar un ensayo preliminar a fin de determinar si es completamente soluble en el reactivo.
Para analizar las mezclas que contienen clorofibras también se puede aplicar el método n° 9 o el método n° 14.
2. PRINCIPIO.
Las fibras de acetato, de triacetato, las clorofibras, determinados modacrílicos y elastanos, se disuelven a partir de un peso conocido de mezcla en su estado seco, por extracción a temperatura cercana a la de ebullición mediante ciclohexanona. El residuo se recogerá, lavado, secado y pesado; su peso, corregido si fuera necesario, se expresará en porcentaje del peso de la mezcla en estado seco. El porcentaje en estado seco de clorofibra, modacrílico, elastano, acetato y triacetato se obtendrá por diferencia.
3. INSTRUMENTAL Y REACTIVOS (además de los mencionados en las generalidades).
3.1. Instrumental.
i) Aparato para la extracción en caliente que permita seguir el modo de operar previsto en el punto 4 [ver el croquis variante del instrumental descrito en Melliand Textilberichte 56 (1975) pp. 643-645].
ii) Placa filtrante en la que se colocará la muestra.
iii) Placa porosa, porosidad 1.
iv) Refrigerante de reflujo adaptable al matraz de destilación.
v) Aparato térmico.
3.2. Reactivos.
i) Ciclohexanona, punto de ebullición a 156 ºC.
ii) Alcohol etílico diluido a un 50 % del volumen.
Nota:
La ciclohexanona es inflamable y tóxica; al utilizarla deben tomarse medidas de protección adecuadas.
4. MODO DE OPERAR.
Seguir las instrucciones facilitadas en las generalidades y proceder de la manera siguiente:
Verter en el matraz de destilación 100 ml de ciclohexanona por gramo de materia, insertar el recipiente de extracción en el que se habrán colocado previamente la placa filtrante con la muestra y la placa porosa que se mantendrá ligeramente inclinada. Introducir el refrigerante de reflujo. Llevar a ebullición y continuar la extracción durante 60 minutos a una velocidad mínima de 12 ciclos por hora. Después de la extracción y del enfriamiento, retirar el recipiente de extracción, sacar la placa filtrante y retirar la placa porosa. Lavar 3 ó 4 veces el contenido de la placa filtrante con alcohol etílico al 50 % precalentado a unos 60 ºC y después con 1 l de agua a 60 ºC.
Durante y entre cada uno de los lavados, no debe aplicarse el vacío, sino que deberá dejarse que el disolvente se vacíe por gravedad y aplicar a continuación el vacío.
Secar la placa con el residuo, enfriar y pesar.
5. CÁLCULO Y PRESENTACIÓN DE LOS RESULTADOS.
Calcular los resultados de la manera descrita en las generalidades. El valor de «d» es de 1,00, excepto para:
La seda 1,01.
El acrílico 0,98.
6. PRECISIÓN DEL MÉTODO.
En mezclas homogéneas de materias textiles, los márgenes de fiabilidad de los resultados obtenidos mediante este método no son superiores a ± 1, para un margen de fiabilidad del 95 %.
____________
Croquis contemplado en el punto 3.1.i) del método n.º 15
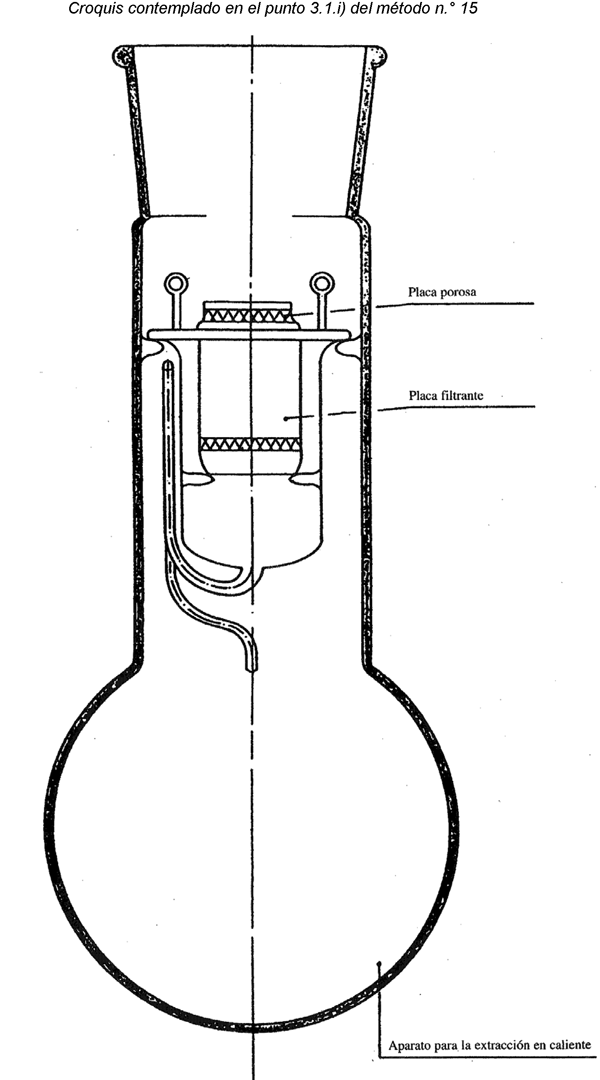
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




