Agencia Estatal Boletín Oficial del Estado






El organismo nocivo, Clavibacter michiganensis (Smith) Davis et al. ssp. sepedonicus, es la causa de la enfermedad denominada necrosis bacteriana de la patata y representa una grave amenaza para las producciones de estos tubérculos.
La Directiva 93/85/CEE del Consejo, de 4 de octubre de 1993, relativa a la lucha contra la necrosis bacteriana de la patata, establece las medidas detalladas a adoptar en el seno de la Comunidad para la lucha contra este organismo nocivo, y fue incorporada al ordenamiento jurídico español por la Orden de 22 de marzo de 1994, relativa a la lucha contra la necrosis bacteriana de la patata en aplicación de la Directiva 93/85/CEE del Consejo de las Comunidades Europeas. Dichas medidas tienen como finalidad localizar el organismo y determinar su distribución, impedir su aparición y, en caso de que esta tenga lugar, evitar su propagación y combatirlo con el fin de erradicarlo.
Teniendo en cuenta que en los últimos años el conocimiento de esta enfermedad y sus métodos de detección e identificación han avanzado considerablemente y que la experiencia adquirida en el seguimiento y lucha contra este organismo aconsejan la revisión de determinadas disposiciones técnicas relacionadas con las medidas de control, se han modificado los anexos de la mencionada directiva por la Directiva 2006/56/CE de la Comisión, de 12 de junio de 2006, por la que se modifican los anexos de la Directiva 93/85/CEE del Consejo, relativa a la lucha contra la necrosis bacteriana de la patata.
En consecuencia, mediante esta Orden se incorpora al ordenamiento interno la Directiva 2006/56/CE de la Comisión, de 12 de junio de 2006.
En la elaboración de la presente disposición han sido consultadas las comunidades autónomas y las entidades representativas de los sectores afectados.
En su virtud, dispongo:
Los anexos de la Orden de 22 de marzo de 1994, relativa a la lucha contra la necrosis bacteriana de la patata en aplicación de la Directiva 93/85/CEE del Consejo de las Comunidades Europeas, se sustituyen por los que figuran como anexo de la presente Orden.
La presente orden se dicta al amparo del artículo 149.1.13.a de la Constitución, que atribuye al Estado la competencia exclusiva sobre las bases y coordinación de la planificación general de la actividad económica.
La presente orden entrará en vigor el día 1 de abril de 2007.
Madrid, 15 de marzo de 2007.‒La Ministra de Agricultura, Pesca y Alimentación, Elena Espinosa Mangana.
ÁMBITO DE APLICACIÓN DEL MÉTODO
El método expuesto describe diversos procedimientos relacionados con:
i) el diagnóstico de la necrosis bacteriana en tubérculos y plantas de patata,
ii) la detección de Clavibacter michiganensis subsp. sepedonicus en muestras de tubérculos y plantas de patata,
iii) la identificación de Clavibacter michiganensis subsp. sepedonicus (C. m. subsp. sepedonicus).
PRINCIPIOS GENERALES
Los apéndices recogen protocolos optimizados para los distintos métodos, reactivos validados y datos relativos a la preparación de los materiales utilizados en las pruebas y de los materiales de control. El apéndice 1 incluye una lista de los laboratorios que participaron en la optimización y la validación de los protocolos.
Dado que los protocolos entrañan la detección de un organismo sujeto a cuarentena e incluirán la utilización de cultivos viables de C. m. subsp. sepedonicus como materiales de control, será necesario ejecutar los procedimientos en las condiciones de cuarentena apropiadas con las instalaciones necesarias para el desecho de los residuos y contando con los permisos pertinentes emitidos por las autoridades oficiales responsables de la cuarentena fitosanitaria.
Los parámetros de prueba deben garantizar la detección coherente y reproducible de los niveles de C. m. subsp. sepedonicus en los límites fijados por los métodos seleccionados.
Es esencial una preparación precisa de los controles positivos.
El hecho de ejecutar las pruebas de acuerdo con los límites exigidos implica también la utilización de los ajustes correctos, el mantenimiento y la calibración del equipo, una cuidadosa manipulación y conservación de los reactivos, y la aplicación de todas las medidas destinadas a evitar la contaminación entre las muestras, como, por ejemplo, la separación de los controles positivos de las muestras de prueba. Se deben aplicar normas relativas al control de la calidad con el fin de evitar errores administrativos y de otro tipo, especialmente por lo que se refiere al etiquetado y la documentación.
Toda aparición sospechosa, como la referida en el artículo 4, apartado 2, de esta orden, entraña un resultado positivo en las pruebas de diagnóstico o de selección realizadas con una muestra tal como se especifica en los diagramas de flujo.
Si en la primera prueba de selección (IF o PCR/FISH) se obtiene un resultado positivo, puede sospecharse la contaminación con C. m. subsp. sepedonicus y se debe efectuar una segunda prueba de selección. Si el resultado de la segunda prueba de selección es positivo, la sospecha se confirma (aparición sospechosa) y las pruebas deben proseguir de acuerdo con el método. En el caso de que el resultado de la segunda prueba de selección sea negativo, la muestra no se considerará contaminada con C. m. subsp. sepedonicus.
Por tanto, los resultados positivos de una prueba IF a los que hace referencia el artículo 4, apartado 2, se definen mediante una lectura positiva en una prueba IF confirmada por una segunda prueba de selección (PCR/FISH).
La presencia confirmada a la que se hace referencia en el artículo 5, apartado 1 de esta orden, requiere el aislamiento y la identificación de un cultivo puro de C. m. subsp. sepedonicus con confirmación de su patogenicidad.
1. PRESENTACIÓN EN DIAGRAMAS DE FLUJO.
1.1 Método de detección para el diagnóstico de la necrosis bacteriana en tubérculos y plantas de patata que presentan síntomas de necrosis bacteriana.
Este protocolo de análisis está destinado a los tubérculos y plantas de patata que muestran síntomas típicos o sospechosos de necrosis bacteriana. Incluye una prueba de selección rápida, el aislamiento del patógeno del tejido vascular infectado en un medio de diagnóstico y, en caso de resultado positivo, la identificación del cultivo como C. m. subsp. Sepedonicus.
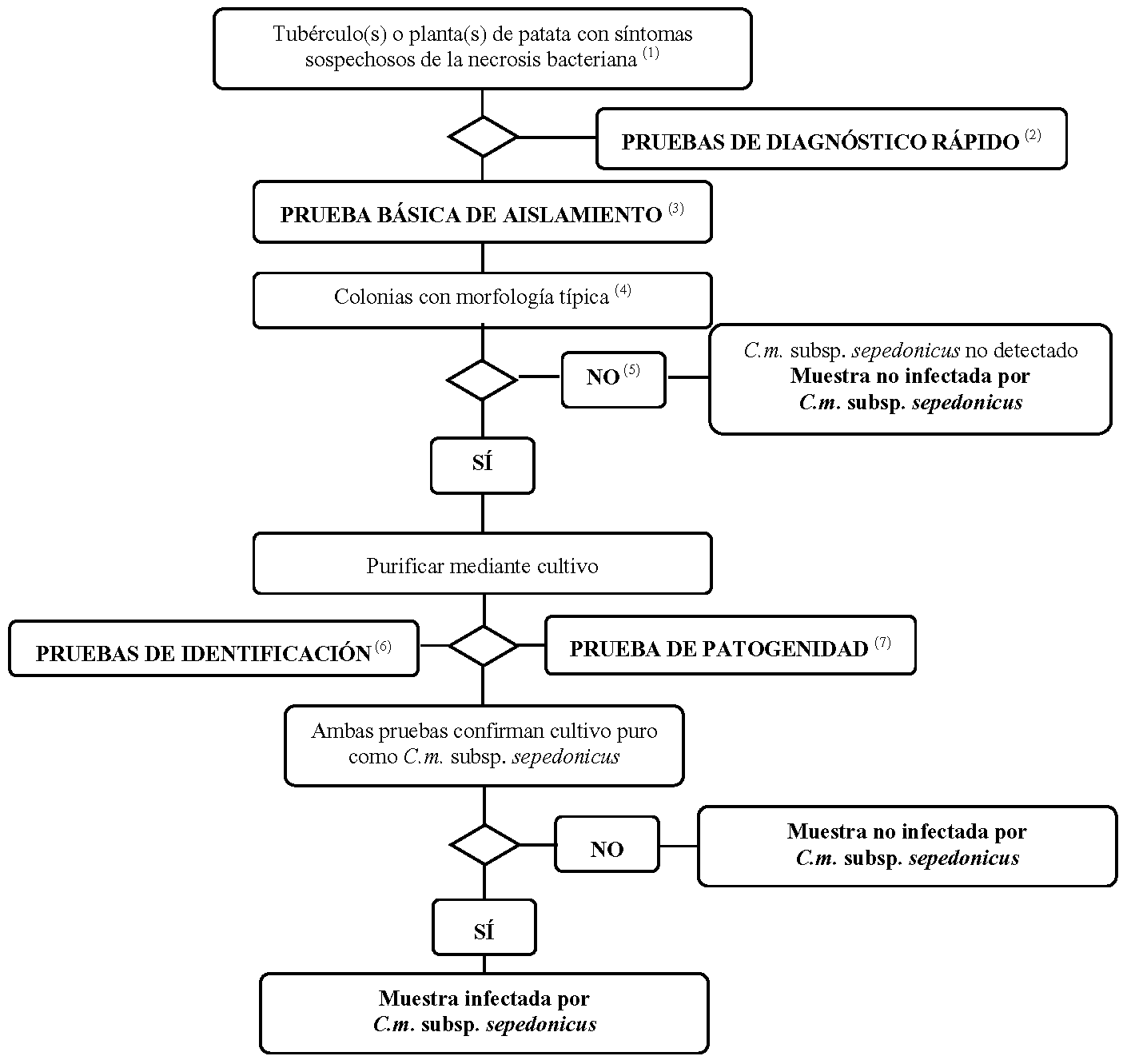
(1) El punto 2 recoge una descripción de los síntomas.
(2) Se consideran adecuadas las pruebas siguientes:
– la prueba IF (punto 4),
– la prueba PCR (punto 5),
– la prueba FISH (punto 6).
(3) Aunque el aislamiento de patógeno a partir de material vegetal con síntomas típicos mediante dilución de placas es sencillo, el aislamiento puede fallar en estados avanzados de infección, las bacterias saprofitas que crecen en el tejido enfermo pueden enmascarar o inhibir el patógeno en el medio de aislamiento. Se recomienda, por tanto, utilizar medios selectivos y no selectivos, preferiblemente MTNA (punto 8) o la prueba de bioensayo (punto 7).
(4) El punto 8 recoge una descripción de la morfología típica de las colonias.
(5) Si los resultados de la prueba de aislamiento son negativos, pero los síntomas de la enfermedad son los típicos, deberá repetirse el aislamiento.
(6) La identificación fiable de un cultivo puro de C.m. subsp. sepedonicus se consigue utilizando las pruebas que se enumeran en el punto 9.
(7) En el punto 10 se describe la prueba de patogenicidad.
1.2 Método de detección e identificación de Clavibacter michiganensis ssp. sepedonicus en muestras de tubérculos de patata asintomáticos.
Principio.
Este protocolo de análisis se destina a la detección de infecciones latentes en tubérculos de patata asintomáticos mediante al menos dos pruebas de selección basadas en distintos principios biológicos que, de ser positivas, deberán complementarse con el aislamiento del patógeno; en caso de aislamiento de colonias típicas, se procede a continuación a la identificación de un cultivo puro como C. m. subsp. sepedonicus. El hecho de obtener resultados positivos sólo en una de las pruebas no es suficiente para considerar la muestra sospechosa.
Las pruebas de selección y aislamiento deben permitir límites de detección de 103 a 104 células por ml de precipitado resuspendido, incluidos como controles positivos en cada serie de pruebas.
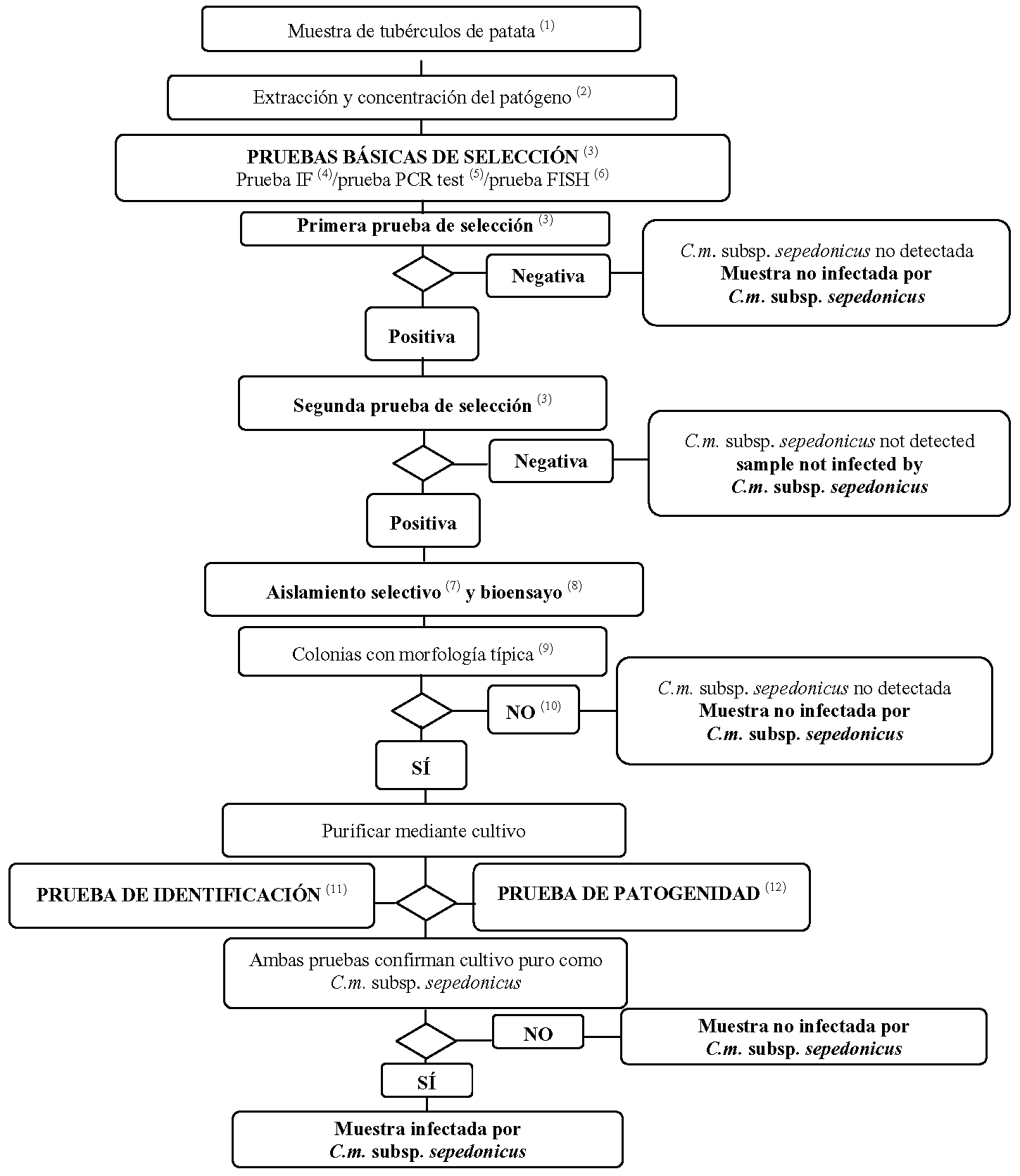
(1) El tamaño normal de la muestra es de 200 tubérculos, aunque el procedimiento puede adaptarse para muestras con menos tubérculos en el caso de que no se disponga de 200.
(2) Los métodos de extracción y concentración del patógeno se describen en el punto 3.1.
(3) En el caso de que al menos dos de las pruebas basadas en principios biológicos diferentes proporcionen resultados positivos, habrá de procederse al aislamiento y la confirmación. Debe realizarse como mínimo una prueba de selección. Cuando el resultado de esta prueba sea negativo, la muestra se considerará negativa. En el caso de que el resultado de esta prueba sea positivo, será necesario realizar dos o más pruebas de selección basadas en principios biológicos diferentes con el fin de verificar el primer resultado positivo. Sí la segunda prueba o el resto de las pruebas proporcionan resultados negativos, la muestra se considerará negativa y no será necesario llevar a cabo más pruebas.
(4) Prueba de inmunofluorescencia (IF}. Para la prueba de selección IF debe utilizarse siempre un anticuerpo policional, los anticuerpos monoclonales adicionales pueden proporcionar una mayor especificidad (véase el punto 4).
(5) Prueba PCR. Deben utilizarse reactivos y protocolos PCR convenientemente validados (véase el punto 6).
(6) Prueba FISH. Deben utilizarse reactivos y protocolos convenientemente validados (véase el punto 5).
(7) Aislamiento selectivo. Con el medio MTNA o el medio NCP-88 y una dilución al 1/100 del precipitado resuspendido, es en muchos casos un método adecuado para el aislamiento directo de C. m. subsp. sepedonicus. Entre 3 y 10dias después de la siembra selectiva pueden obtenerse colonias típicas. Posteriormente se puede purificar e identificar el patógeno. Para poder aprovechar plenamente el potencial de la prueba, es necesario preparar con cuidado las cuñas de la parte basal a fin de evitar que otras bacterias secundarias asociadas al tubérculo de la patata, que compitan con C. m. subsp. sepedonicus en el medio, puedan afectar al desarrollo del patógeno. Si la prueba de la siembra selectiva no resulta efectiva, se deberá proceder al aislamiento a partir de las plantas utilizadas para el bioensayo (véase el punto 8).
(8) La prueba de bioensayo se utiliza para el aislamiento de C. m. subsp. sepedonicus a partir de precipitado de extracto de patatas mediante el enriquecimiento selectivo de la bacteria en berenjenas (Solanum melongena). Exige que las condiciones de incubación sean óptimas, según se especifican en este método. Es muy probable que las bacterias inhibidoras de C. m. subsp. sepedonicus en los medios MTNA o NCP-88 no interfieran en esta prueba (véase el punto 7).
(9) En el punto 8 se describe la morfología típica de las colonias.
(10) Los cultivos o bioensayos pueden fallar debido a la competencia de las bacterias saprofitas o a la inhibición provocada por las mismas. En el caso de que se obtengan resultados positivos en las pruebas de selección pero negativos en las de aislamiento, se deberán repetir las pruebas de aislamiento a partir del mismo precipitado o utilizando además tejido vascular de la parte basal de tubérculos cortados de la misma muestra y, si fuese necesario, con otras muestras.
(11) La identificación fiable de cultivos puros supuestamente de C. m. subsp. sepedonicus se consigue utilizando las pruebas que se describen en el punto 9.
(12) En el punto 10 se describe la prueba de patogenicidad.
1.3 Método de detección e identificación de Clavibacter michiganensis ssp. sepedonicus en muestras de plantas de patata asintomáticas.
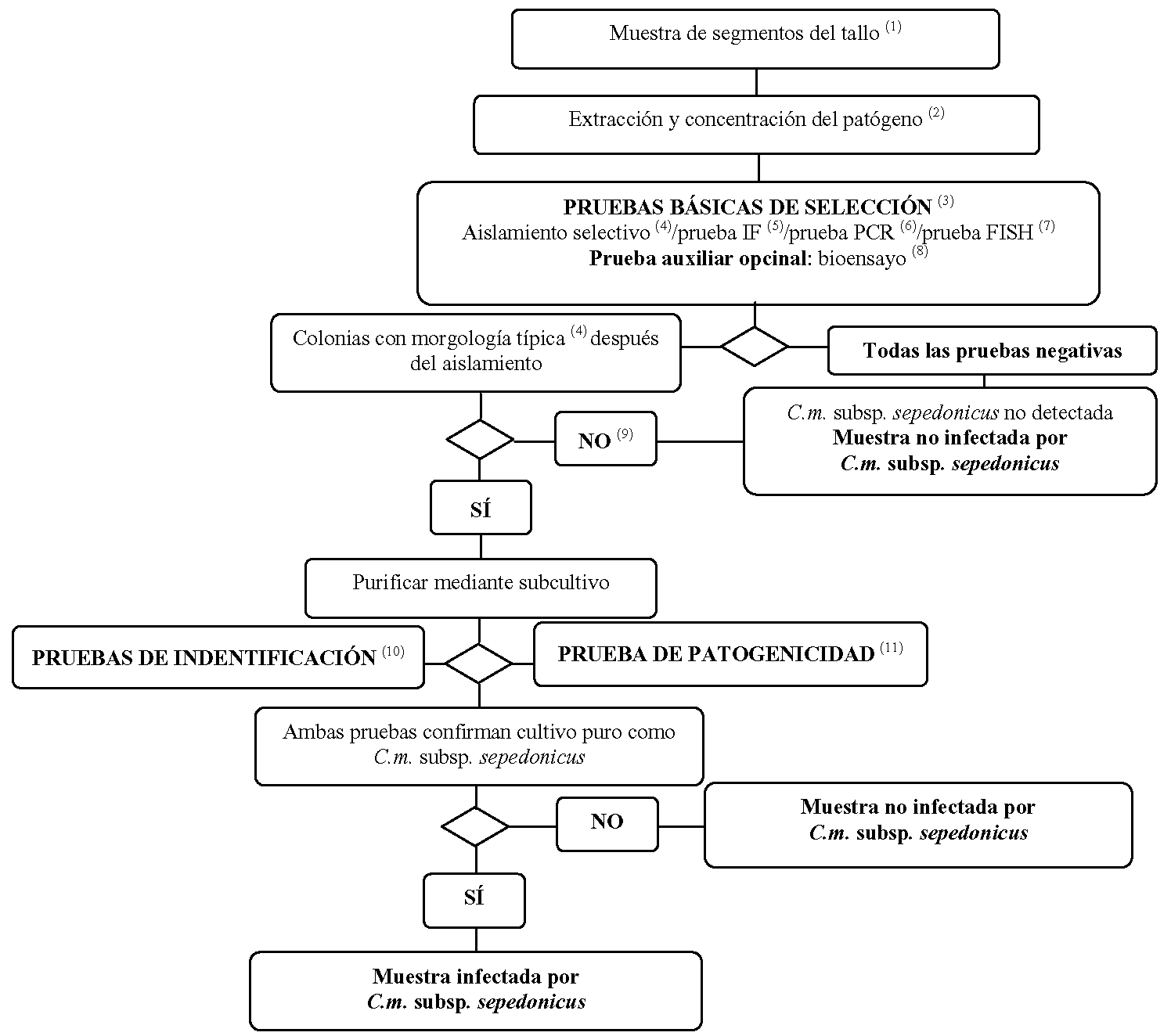
(1) Véase el punto 3.2 para obtener información sobre los tamaños recomendados de la muestra.
(2) Los métodos de extracción y concentración del patógeno se describen en el punto 3.2.
(3) En el caso de que al menos dos de las pruebas basadas en principios biológicos diferentes proporcionen resultados positivos, habrá de procederse al aislamiento y la confirmación. Debe realizarse como mínimo una prueba de selección. Cuando el resultado de la prueba sea negativo, la muestra se considerará negativa. En el caso de que el resultado de esta prueba sea positivo, será necesario realizar dos o más pruebas de selección basadas en principios biológicos diferentes con el fin de verificar el primer resultado positivo. Si la segunda prueba o el resto de las pruebas proporcionan resultados negativos, la muestra se considerará negativa y no será necesario llevar a cabo más pruebas.
(4) En el punto 8 se describe la prueba de aislamiento selectivo y la morfología típica de las colonias.
(5) En el punto 4 se describe la prueba IF.
(6) En el punto 6 se describe la prueba PCR.
(7) En el punto 5 se describe la prueba FISH.
(8) En el punto 7 se describe la prueba de bioensayo.
(9) Los cultivos o bioensayos pueden fallar debido a la competencia de las bacterias saprofitas o a la inhibición provocada por las mismas. En el caso de que se obtengan resultados positivos en las pruebas de selección pero negativos en las de aislamiento, se deberán repetir las pruebas de aislamiento y, si fuese necesario, efectuar pruebas con otras muestras.
(10) La identificación fiable de cultivos puros supuestamente de C. m. subsp. sepedonicus se consigue utilizando las pruebas que se describen en el punto 9.
(11) En el punto 10 se describe la prueba de patogenicidad.
2. EXAMEN VISUAL PARA DETECTAR SÍNTOMAS DE NECROSIS BACTERIANA.
2.1 Plantas de patata.
En las condiciones climáticas europeas, los síntomas se presentan rara vez en el campo y con frecuencia sólo al final de la temporada. Por otra parte, en numerosos casos, otras enfermedades, la senescencia o los daños mecánicos enmascaran los síntomas o se confunden con ellos. Por tanto, puede ser fácil pasar por alto los síntomas en las inspecciones de campo. Los síntomas de marchitamiento son muy distintos de los de la podredumbre parda; el marchitamiento es normalmente un proceso lento y se limita en principio a los bordes de las hojas. El crecimiento de las hojas jóvenes infectadas no suele detenerse, aunque sea más lento en las zonas infectadas. Esto hace que las hojas tengan formas extrañas. Las hojas afectadas por el bloqueo de los tejidos vasculares de la parte baja del tallo desarrollan con frecuencia zonas intenerviales cloróticas amarillas o anaranjadas. Los foliolos, hojas e incluso tallos infectados pueden llegar a morir. Con frecuencia las hojas y tubérculos muestran simplemente un tamaño reducido. En algunos casos las plantas se atrofian. Se pueden consultar fotografías en color de algunos de estos síntomas en el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main
2.2 Tubérculos de patata.
Los primeros síntomas consisten en un ligero aspecto vítreo o traslúcido del tejido, sin ablandamiento, en torno al sistema vascular, en especial cerca de la parte basal. La corona vascular de la parte basal puede tener un color ligeramente más oscuro de lo normal. El primer síntoma claramente detectable consiste en que la corona vascular adquiere una coloración amarillenta y al comprimir suavemente el tubérculo emergen de los vasos unos exudados de aspecto lechoso. Dicho exudado contiene millones de bacterias. Se puede producir el oscurecimiento del tejido vascular y los síntomas que presentan los tubérculos en esta fase son similares a los de la podredumbre parda provocada por Ralstonia solanacearum. Al principio, estos síntomas pueden limitarse a una parte de la corona, no necesariamente cerca de la parte basal, y pueden extenderse posteriormente a la totalidad de la corona.
A medida que la infección progresa, se produce destrucción de tejido vascular; la zona cortical externa puede separarse de la zona cortical interna. En las fases avanzadas de la infección, se producen grietas en la superficie del tubérculo, que suelen tener un color pardo rojizo en los bordes. Recientemente se han producido varios casos en Europa en los que la zona cortical central se ha podrido al mismo tiempo que la corona vascular provocando una invasión secundaria con oquedades y necrosis interna. Los síntomas pueden verse enmascarados por una invasión secundaria fúngica o bacteriana que puede hacer difícil, o incluso imposible, diferenciar los síntomas de necrosis bacteriana avanzada de otras necrosis de los tubérculos. Pueden presentarse síntomas atípicos. Se pueden consultar fotografías en color de algunos de estos síntomas en el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main
3 PREPARACIÓN DE LA MUESTRA.
3.1 Tubérculos de patata.
Nota:
‒ El tamaño normal de la muestra es de 200 tubérculos por ensayo. Para realizar un muestreo más intensivo es preciso llevar a cabo más pruebas con muestras de este tamaño. El hecho de disponer de un número mayor de tubérculos en la muestra puede provocar la inhibición o dificultar la interpretación de los resultados. No obstante, el procedimiento puede adaptarse convenientemente a muestras formadas por un número menor de tubérculos en el caso de que no se disponga de 200.
‒ La validación de todos los métodos de detección descritos más adelante se basa en los ensayos realizados con muestras de 200 tubérculos.
‒ El extracto de patata descrito a continuación puede emplearse también para la detección de la bacteria de la podredumbre parda de la patata, Ralstonia solanacearum.
Pretratamiento opcional previo a la preparación de la muestra:
Lavar los tubérculos. Utilizar desinfectantes (cuando se vaya a efectuar la prueba PCR, la desinfección con compuestos de cloro puede interferir en la extracción de ADN del patógeno) y detergentes adecuados para cada muestra. Secar los tubérculos al aire. Este procedimiento de lavado resulta particularmente útil (aunque no necesario) en el caso de muestras con exceso de tierra y cuando se vaya a efectuar una prueba PCR o un procedimiento de aislamiento directo.
3.1.1 Quitar la epidermis de la parte basal de cada tubérculo con un bisturí o un cuchillo limpio y desinfectado, de modo que el tejido vascular quede a la vista. Extraer cuidadosamente una pequeña cuña de tejido vascular de la parte basal y el mínimo volumen posible de tejido no vascular (véase el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main)
Nota: Retirar los tubérculos que presenten síntomas sospechosos de necrosis bacteriana y analizarlos separadamente.
En el caso de que se observen síntomas sospechosos de necrosis bacteriana durante la extracción de la cuña de la parte basal, deberá realizarse una inspección visual del tubérculo una vez cortado a la altura de la parte basal.
Todo tubérculo cortado que presente síntomas sospechosos deberá suberizarse a temperatura ambiente durante dos días y almacenarse en cuarentena (a una temperatura de entre 4 y 10 °C) hasta que concluyan todos los ensayos. Todas las muestras de tubérculos, incluidas las que presenten síntomas sospechosos, deberán conservarse de acuerdo con lo dispuesto en el anexo II.
3.1.2 Introducir las cuñas de la parte basal en recipientes desechables que no hayan sido utilizados previamente y que puedan cerrarse y/o sellarse (en el caso de que estos recipientes vayan a volver a utilizarse deberán limpiarse y desinfectarse concienzudamente con compuestos de cloro). Preferiblemente, las cuñas deben procesarse inmediatamente. Si esto no fuese posible, deberán almacenarse en el recipiente, sin tampón, y conservarse por un período máximo de 72 horas refrigeradas o de 24 horas a temperatura ambiente. El secado y la suberización de las cuñas, así como el crecimiento de saprofitos durante el almacenamiento, pueden dificultar la detección de la bacteria causante de la necrosis bacteriana.
3.1.3 Procesar las cuñas básales por uno de los métodos siguientes:
a) bien añadir la solución de tampón de extracción (apéndice 3) en cantidad suficiente para que cubra las cuñas (aproximadamente 40 ml) y agitar en un agitador rotatorio (50-100 rpm) durante 4 horas a una temperatura inferior a 24 °C o durante 16 a 24 horas si están refrigeradas;
b) o bien homogeneizar las cuñas con solución de tampón de extracción (apéndice 3) en cantidad suficiente (aproximadamente 40 ml), ya sea mediante una trituradora (por ejemplo, Waring o Ultra Thurax) o machacándolas en una bolsa de maceración desechable sellada (por ejemplo, bolsas resistentes Stomacher o Bioreba de polietileno de 150 mm × 250 mm, esterilizadas por radiación) utilizando un mazo de caucho o un aparato triturador adecuado (por ejemplo, Homex).
Nota: El riesgo de contaminación cruzada de las muestras es alto cuando éstas se homogeneizan mediante una trituradora. Se debe actuar con precaución para evitar la generación de aerosoles o el vertido durante el proceso de extracción. Es preciso asegurarse de que se esterilizan las cuchillas de la trituradora y los recipientes que vayan a utilizarse para cada muestra. Si se va a realizar la prueba PCR, es preciso evitar que existan restos de ADN en los recipientes o el aparato triturador. Cuando se vaya a efectuar la prueba PCR se recomienda la trituración en bolsas desechables y la utilización de tubos desechables.
3.1.4 Decantar el sobrenadante. Si presenta un aspecto excesivamente turbio, clarificarlo centrifugándolo a baja velocidad (a 180 g como máximo, durante 10 minutos, a una temperatura entre 4 y 10 °C) o mediante filtración al vacío (40-100 μm), lavando el filtro con solución adicional (10 ml) de tampón de extracción (apéndice 3).
3.1.5 Concentrar la fracción bacteriana mediante centrifugación a 7.000 g durante 15 minutos (o a 10.000 g durante 10 minutos) a una temperatura entre 4 y 10 °C y descartar el sobrenadante sin perturbar el precipitado.
3.1.6 Resuspender el precipitado en un 1,5 ml de tampón de precipitado (apéndice 3). Utilizar 500 μl para la prueba de C. m. subsp. sepedonicus, 500 μl para Ralstonia solanacearum y 500 μl a efectos de referencia. Añadir glicerol estéril en una concentración final de 10-25 % (v/v) a los 500 μl de la alícuota de referencia y a la alícuota restante de cada prueba, homogeneizar por agitación y guardar a una temperatura entre ‒16 y ‒24 °C (semanas) o entre ‒68 y ‒86 °C (meses). Mantener las alícuotas de la prueba a una temperatura comprendida entre 4 y 10 °C durante los ensayos.
No es recomendable congelar y descongelar el extracto en repetidas ocasiones.
Si fuese preciso transportar el extracto, es conveniente asegurarse de que la entrega se efectúa en una caja refrigerada en el plazo de 24 a 48 horas.
3.1.7 Es imprescindible que todos los controles y muestras positivas de C. m. subsp. sepedonicus se procesen por separado, a fin de evitar la contaminación. Esto se aplica a las preparaciones para IF y a todas las pruebas.
3.2 Plantas de patata.
Nota: Para la detección de poblaciones latentes de C. m. subsp. sepedonicus se recomienda efectuar las pruebas con muestras mixtas. El procedimiento se puede adaptar convenientemente para muestras mixtas de hasta 200 partes de tallos (los estudios deberán basarse en una muestra representativa desde el punto de vista estadístico de la población vegetal que se someta a investigación).
3.2.1 Extraer una sección de 1 a 2 cm de la base de cada tallo, justo por encima del nivel del suelo, utilizando un cuchillo o tijeras de podar limpios y desinfectados.
Desinfectar ligeramente las secciones de tallo con etanol al 70 % y secar inmediatamente con un pañuelo de papel.
Introducir las secciones de tallo en un recipiente estéril cerrado de acuerdo con los siguientes procedimientos de muestreo:
3.2.2 Procesar las secciones de tallo mediante uno de los métodos siguientes:
a) bien añadir la solución de tampón de extracción (apéndice 3) en cantidad suficiente para que cubra las secciones (aproximadamente 40 ml) y agitar en un agitador rotatorio (50-100 rpm) durante 4 horas a una temperatura inferior a 24 °C o durante 16 a 24 horas si están refrigeradas;
b) o bien procesar inmediatamente triturando las secciones en una bolsa de maceración resistente (p. ej. Stomacher o Bioreba) con una cantidad adecuada de tampón de extracción (apéndice 3) utilizando para ello un mazo de caucho o un aparato triturador adecuado (p. ej. Homex). Si esto no es posible, almacenar las secciones de tallo refrigeradas durante 72 horas como máximo o a temperatura ambiente durante 24 horas como máximo.
3.2.3 Decantar el sobrenadante tras haberlo dejado reposar durante 15 minutos.
3.2.4 Normalmente no es necesario clarificar más el extracto ni la concentración de la fracción bacteriana, pero esto se puede lograr mediante la filtración y/o la centrifugación, tal como se describe en los puntos 3.1.4 a 3.1.6.
3.2.5 Dividir el extracto puro o concentrado de la muestra en dos partes iguales. Mantener una de las partes a 4-10 °C durante los ensayos y almacenar la parte restante, tras haber añadido glicerol estéril al 10-25 % (v/v), a una temperatura comprendida entre ‒16 y ‒24 °C (semanas) o entre ‒68 y ‒86 °C (meses) en el caso de que sea necesario realizar nuevas pruebas.
4. PRUEBA IF.
Principio:
La utilización de la prueba IF como principal prueba de selección está recomendada por su solidez demostrada para alcanzar los límites exigidos.
Cuando se utilice la prueba IF como principal prueba de selección y ésta proporcione resultados positivos, deberán realizarse las pruebas PCR o FISH como segunda prueba de selección. Cuando se utilice la prueba IF como segunda prueba de selección y ésta proporcione resultados positivos, será preciso realizar las pruebas establecidas en el diagrama de flujo para completar el análisis.
Nota: Cuando se utilice la prueba IF como principal prueba de selección, deberá utilizarse siempre un anticuerpo policlonal. En el caso de resultados IF positivos con un anticuerpo policlonal, la selección posterior de la muestra con un anticuerpo monoclonal puede proporcionar una mayor especificidad pero ser menos sensible.
Conviene utilizar anticuerpos frente a la cepa de referencia de C. m. subsp. sepedonicus. Se recomienda determinar el título para cada nuevo lote de anticuerpos. El título se define como la dilución superior en la que se produce la reacción óptima cuando se realiza la prueba con una suspensión que contiene entre 105 y 106 células por ml de la cepa homologa de C. m. subsp. sepedonicus y se utiliza una dilución apropiada del conjugado de isotiocianato de fluoresceína (FITC), según las recomendaciones del fabricante.
Los anticuerpos policlonales o monoclonales crudos deberían tener un título IF de al menos 1:2000. Durante las pruebas, los anticuerpos deberían utilizarse en diluciones de trabajo (DT) próximas o iguales al título. Utilizar anticuerpos validados.
(véase el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main)
La prueba debería realizarse con extractos de muestras que se acaben de preparar. Si fuese necesario, puede efectuarse también con extractos almacenados a una temperatura comprendida entre ‒68 y ‒86 °C en glicerol. El glicerol puede separarse de la muestra mediante la adición de 1 ml de tampón de precipitado (apéndice 4), la recentrifugación durante 15 minutos a 7.000 g y la resuspensión en el mismo volumen de tampón de precipitado. Normalmente esto no es necesario, especialmente si las muestras se fijan al portaobjetos flameándolas (véase el punto 2.2).
Preparar otros portaobjetos con controles positivos de la cepa homologa o de cualquier otra cepa de referencia de C. m. subsp. sepedonicus, suspendida en extracto de patata, tal como se especifica en el apéndice 2 y, opcionalmente, en tampón.
De ser posible, deberán utilizarse tejidos infectados de forma natural (conservados mediante liofilización o congelación a temperaturas comprendidas entre ‒16 y ‒24 °C) como control similar, en el mismo portaobjetos.
Como controles negativos, utilizar alícuotas de extractos de muestra que previamente hayan proporcionado resultados negativos en la prueba.
Utilizar portaobjetos de pocillos múltiples, de preferencia con 10 pocillos de 6 mm de diámetro como mínimo.
Procesar el material de control de la misma manera que la muestra o muestras.
4.1 Preparar los portaobjetos según uno de los procedimientos siguientes:
i) Para precipitados con una cantidad relativamente pequeña de sedimento de almidón:
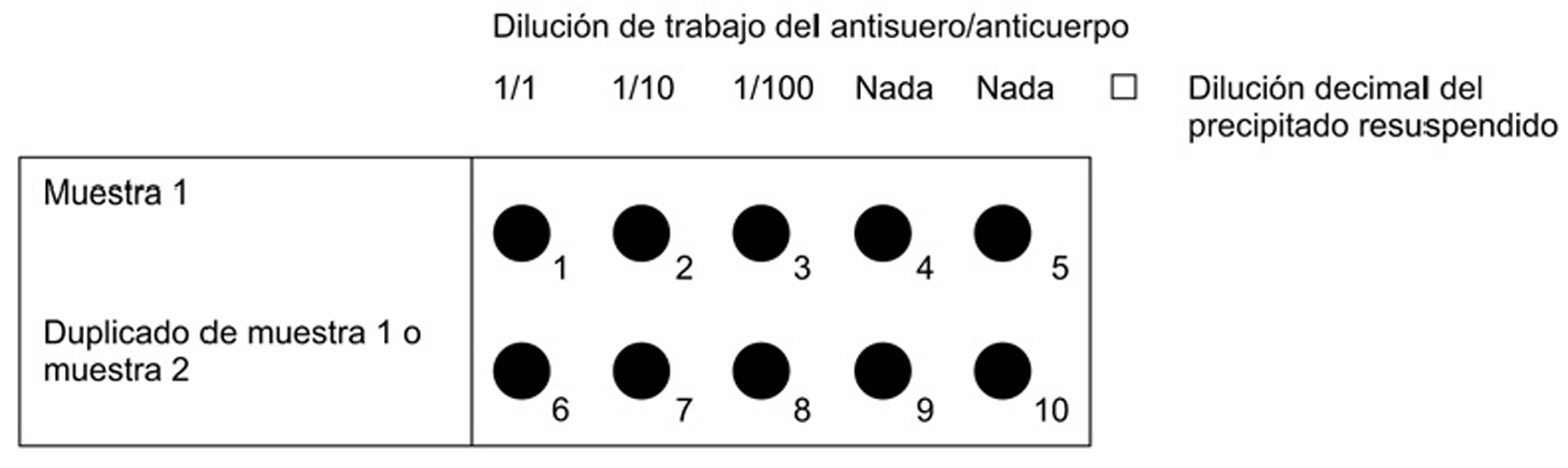
Verter con una pipeta un volumen determinado (15 μl es suficiente para pocillos de 6 mm de diámetro; aumentar el volumen si el diámetro es mayor) de una dilución al 1/100 del precipitado de patata resuspendido en el primer pocillo. Posteriormente, verter un volumen similar de precipitado sin diluir (1/1) en los demás pocillos de la fila. La otra fila puede utilizarse como duplicado o para una segunda muestra, tal como se indica en la figura 1.
ii) Para otros precipitados:
Preparar diluciones decimales (1/10 y 1/100) del precipitado resuspendido en el tampón de precipitado. Verter con una pipeta un volumen determinado (15 μl es suficiente para pocillos de 6 mm de diámetro; aumentar el volumen si el diámetro es mayor) del precipitado resuspendido y de cada dilución en una de las filas de pocillos. La otra fila puede utilizarse como duplicado o para una segunda muestra, tal como se indica en la figura 2.
4.2 Dejar secar las gotitas a temperatura ambiente o calentándolas a una temperatura comprendida entre 40 y 45 °C.
Fijar las células bacterianas al portaobjetos calentándolo (15 minutos a 60 °C), flameándolo, mediante etanol al 95 %, o de acuerdo con las instrucciones específicas de los proveedores de los anticuerpos.
De ser necesario, los portaobjetos fijados pueden almacenarse congelados en una caja seca durante el mínimo tiempo posible (hasta un máximo de tres meses) antes de realizar nuevas pruebas.
4.3 Procedimiento IF:
i) si el portaobjetos se ha preparado según el inciso i) del punto 4.1:
Preparar un conjunto de diluciones a 1/2 del anticuerpo en el tampón IF. El primer pocillo deberá tener 1/2 del título (T/2) y el resto 1/4 del título (T/4), 1/2 del título (T/2), el título (T) y dos veces el título (2T),
ii) si el portaobjetos se ha preparado según el inciso ii) del punto 4.1:
Preparar la dilución de trabajo (DT) del anticuerpo en tampón IF. La dilución de trabajo afecta a la especificidad.
Figura 1. Preparación del portaobjetos de acuerdo con el punto 4.1, inciso i), y el punto 4.3, inciso i)
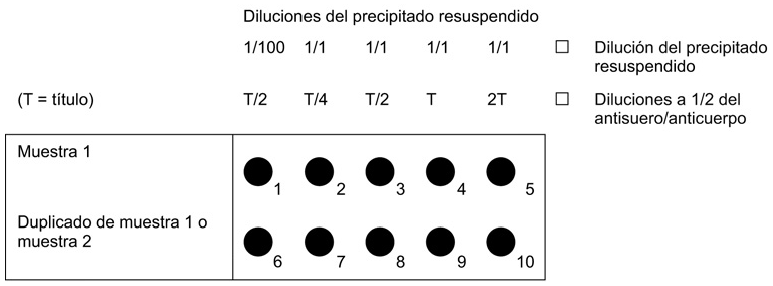
Figura 2. Preparación del portaobjetos de acuerdo con el punto 4.1, inciso ii), y el punto 4.3, inciso ii)
4.3.1 Colocar los portaobjetos en papel humedecido. Cubrir completamente cada pocillo con la dilución o diluciones del anticuerpo. El volumen del anticuerpo aplicado en cada pocillo debe ser equivalente al menos al del extracto aplicado.
A falta de instrucciones específicas de los proveedores de anticuerpos, deberá seguirse el procedimiento siguiente:
4.3.2 Tapar los portaobjetos y dejar incubar sobre papel humedecido durante 30 minutos a temperatura ambiente (18 a 25 °C).
4.3.3 Sacudir las gotitas de cada portaobjetos y enjuagar cuidadosamente con tampón IF. Lavar por inmersión durante 5 minutos en tampón IF-Tween (apéndice 3) y repetir la operación durante 5 minutos en tampón IF. Deben evitarse los aerosoles o la transferencia de gotitas que puedan provocar la contaminación cruzada. Eliminar cuidadosamente el exceso de humedad secándolos ligeramente.
4.3.4 Colocar los portaobjetos en papel humedecido. Cubrir los pocillos con la dilución del conjugado FITC utilizado para determinar el título. El volumen de conjugado aplicado en los pocillos debe ser idéntico al volumen de anticuerpo aplicado.
4.3.5 Tapar los portaobjetos y dejar incubar sobre papel humedecido durante 30 minutos a temperatura ambiente (18 a 25 °C).
4.3.6 Sacudir las gotitas de conjugado del portaobjetos. Enjuagar y lavar como antes (punto 4.3.3). Retirar con cuidado el exceso de humedad.
4.3.7 Verter con una pipeta de 5 a 10 μl de tampón fosfato glicerol de 0,1 M (véase el apéndice 3) o una solución comercial similar que proteja la fluorescencia (anti-fading) en cada pocillo y tapar.
4.4 Lectura de la prueba IF:
4.4.1 Examinar los portaobjetos en un microscopio epifluorescente con filtros adecuados para que se produzca la excitación del FITC, con aceite o agua de inmersión y a 500-1.000 aumentos. Recorrer los pocillos a lo largo de dos diámetros perpendiculares entre sí y alrededor del perímetro. En el caso de muestras que no presenten células o sólo un pequeño número de ellas es preciso observar como mínimo 40 campos microscópicos.
En primer lugar, comprobar el control positivo. Las células deben ser fluorescentes brillantes y estar completamente teñidas al título de anticuerpos determinado o la dilución de trabajo. La prueba IF (punto 4) debe repetirse si la tinción no es correcta.
4.4.2 Comprobar si hay células fluorescentes brillantes con la morfología característica de C. m. subsp. sepedonicus en los pocillos de los portaobjetos (véase el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main).
La intensidad de la fluorescencia debe ser equivalente a la de la cepa de control positivo a la misma dilución del anticuerpo o mejor que ésta. Deberán descartarse las células que presenten una tinción incompleta o cuya fluorescencia sea escasa.
En el caso que se sospeche cualquier posible contaminación, deberá repetirse la prueba. Esto puede ocurrir cuando todos los portaobjetos de un lote presenten células positivas debido a la contaminación del tampón o cuando se encuentren células positivas (fuera de los pocillos) en la superficie del portaobjetos.
4.4.3 Existen varios problemas inherentes a la especificidad de la prueba de inmunofluorescencia. En los precipitados de cuñas básales y secciones de tallo de patata pueden aparecer poblaciones de base de células fluorescentes de morfología atípica y bacterias saprofitas con reacción cruzada cuyo tamaño y morfología sean similares a los de C. m. subsp sepedonicus.
4.4.4 Sólo deben considerarse las células fluorescentes con tamaño y morfología típicos, al título o la dilución de trabajo de los anticuerpos, al igual que en el punto 4.3.
4.4.5 Interpretación de los resultados de la prueba IF:
i) si se encuentran células fluorescentes brillantes con morfología característica, calcular el número medio de células típicas por campo microscópico y el número de células típicas por ml de precipitado resuspendido (apéndice 4).
La prueba IF es positiva para las muestras que presenten al menos 5 × 103 células típicas por ml de precipitado resuspendido. La muestra se considera potencialmente contaminada y es necesario realizar nuevas pruebas,
ii) la prueba IF es negativa para las muestras que presenten menos de 5 × 103 células por ml de precipitado resuspendido y la muestra se considera negativa. No es necesario realizar nuevas pruebas.
5. PRUEBA FISH.
Principio:
Cuando se utilice la prueba FISH como primera prueba de selección y ésta proporcione resultados positivos, deberá realizarse la prueba IF como segunda prueba obligatoria de selección. Cuando se utilice la prueba FISH como segunda prueba de selección y ésta proporcione resultados positivos, será preciso realizar las pruebas establecidas en el diagrama de flujo para completar el diagnóstico.
Nota: Utilizar oligosondas específicas para C. m. subsp. sepedonicus validadas (apéndice 7). Las pruebas preliminares realizadas con este método deberán permitir la detección reproducible de al menos 103-104 células de C. m. subsp. sepedonicus por ml añadidas a los extractos de muestras que previamente proporcionaron resultados negativos.
El procedimiento siguiente debería llevarse a cabo, a ser posible, con extracto de muestra que se acabe de preparar, pero también puede realizarse con extracto de muestra que se haya almacenado en glicerol a una temperatura comprendida entre ‒16 y ‒24 °C o entre ‒68 y ‒86 °C.
Como controles negativos, deben utilizarse alícuotas de extracto de muestra que previamente haya arrojado resultados negativos para C. m. subsp. sepedonicus.
Como controles positivos, deben prepararse suspensiones que contengan de 105 a 106 células por ml de C. m. subsp. sepedonicus (por ejemplo, la cepa NCPPB 4053, o PD 406) en tampón fosfato de concentración 0,01 M de un cultivo de 3 a 5 días (para la preparación véase el apéndice 2). Preparar en otro portaobjetos controles positivos de la cepa homologa o de cualquier otra cepa de referencia de C. m. subsp. sepedonicus, suspendida en extracto de patata, tal como se especifica en el apéndice 2.
El uso de una oligosonda eubacteriana marcada con FITC ofrece un control para el proceso de hibridación, ya que teñirá todas las eubacterias que estén presentes en la muestra.
Procesar el material de control de la misma manera que la muestra o muestras.
5.1 Fijación del extracto de patata.
El protocolo siguiente se basa en Wullings et al., (1998):
5.1.1 Preparar la solución de fijación (véase el apéndice 7).
5.1.2 Verter 100 μl de cada extracto de muestra en un tubo eppendorf y centrifugar durante 8 minutos a 7 000 g.
5.1.3 Quitar el sobrenadante y disolver el precipitado en 500 μl de solución fijadora preparada con menos de 24 horas de antelación. Agitar e incubar de un día para otro a 4 °C.
El etanol al 96 % constituye una solución fijadora alternativa. Para utilizarlo, disolver el precipitado a partir del paso 5.1.2 en 50 μl de tampón fosfato de concentración 0,01 M y 50 μl de etanol al 96 %. Agitar la mezcla e incubar a 4 °C durante 30 a 60 minutos.
5.1.4 Centrifugar durante 8 minutos a 7 000 g, quitar el sobrenadante y resuspender el precipitado en 75 μl de tampón fosfato de concentración 0,01 M (véase el apéndice 3).
5.1.5 Colocar 16 μl de las suspensiones fijadas en un portaobjetos múltiple limpio, tal como muestra la figura 3. Aplicar 2 muestras diferentes por portaobjetos sin diluir y utilizar 10 μl para realizar una dilución al 1:100 (en tampón fosfato de concentración 0,01 M). La solución de la muestra restante (49 μl) puede almacenarse a ‒20 °C tras la adición de 1 volumen de etanol al 96 %. En el caso de que sea necesario repetir la prueba FISH, separar el etanol mediante centrifugación y añadir un volumen equivalente de tampón fosfato de concentración 0,01 M (mezclar mediante agitación).
Figura 3

5.1.6 Secar los portaobjetos al aire (o con secador de portaobjetos a 37 °C) y fijarlos flameándolos.
El procedimiento puede interrumpirse en esta fase, pudiéndose proseguir con la hibridación al día siguiente. Los portaobjetos deben almacenarse secos y sin polvo a temperatura ambiente.
5.2 Prehibridación e hibridación.
5.2.1 Preparar una solución de lisozima que contenga 10 mg de lisozima (Sigma L-6876) en 10 ml de tampón (100 mM Tris-HCI, 50 mM EDTA, pH 8,0). Esta solución puede almacenarse pero sólo debe congelarse y descongelarse una vez. Cubrir todas las muestras bien con aproximadamente 50 μl de solución de lisozima e incubar durante 10 minutos a temperatura ambiente. Sumergir los portaobjetos en agua desmineralizada una única vez y secar con papel de filtro.
Alternativamente, en lugar de lisozima, añadir 50 μl de 40-400 μg ml-1 de proteinasa K en tampón (20 mM Tris-HCI, 2 mM CaCl2, pH 7,4) en cada pocillo e incubar a 37 °C durante 30 minutos.
5.2.2 Deshidratar las células en series de etanol escalonadas al 50 %, 80 % y 96 % durante 1 minuto cada una. Secar las preparaciones en un portaobjetos.
5.2.3 Preparar una cámara de incubación húmeda recubriendo el fondo de una caja hermética con un pañuelo de papel o papel de filtro empapado en 1x hybmix (apéndice 7). Preincubar la caja en el horno de hibridación a 55 °C durante 10 minutos como mínimo.
5.2.4 Preparar la solución de hibridación (apéndice 7) con 45 μl por portaobjeto, y preincubar durante 5 minutos a 55 °C.
5.2.5 Colocar los portaobjetos en una placa caliente a 45 °C y aplicar 10 μl de solución de hibridación en cada uno de los 4 pocillos del (o de los) portaobjetos.
5.2.6 Aplicar 2 cubreobjetos (24 × 24 mm) a cada portaobjeto evitando la entrada de aire. Colocar los portaobjetos en la cámara húmeda precalentada y dejar que se produzca la hibridación de un día para otro a 55 °C en la oscuridad.
5.2.7 Preparar 3 vasos de precipitado que contengan 1 I de agua ultrapura, 1 I de 1x hybmix (334 ml 3x hybmix y 666 ml de agua ultrapura) y 1 I de 1/2x hybmix (167 ml 3x hybmix y 833 ml de agua ultrapura). Preincubarlos al baño maría a 55 °C.
5.2.8 Quitar los cubreobjetos de los portaobjetos y colocar los portaobjetos en un soporte.
5.2.9 Lavar el exceso de sonda mediante incubación durante 15 minutos en el vaso de precipitado con 1x hybmix a 55 °C.
5.2.10 Transferir el portaobjetos a una solución de lavado de hybmix al 1/2 e incubar durante otros 15 minutos.
5.2.11 Sumergir las preparaciones brevemente en agua ultrapura y colocarlas en papel de filtro. Eliminar el exceso de humedad cubriendo la superficie con cuidado con papel de filtro. Verter de 5 a 10 μl de solución de montaje protectora de la fluorescencia (p. ej. Vectashield, Vecta Laboratories, CA, USA o equivalente) en cada pocillo y aplicar un cubreobjetos grande (24 × 60 mm) sobre la totalidad de los portaobjetos.
5.3 Lectura de la prueba FISH:
5.3.1 Los portaobjetos deben examinarse inmediatamente utilizando un microscopio epifluorescente a 630 o 1.000 aumentos con aceite de inmersión. Con un filtro adecuado para isotiocianato de fluoresceína (FITC) las células eubacterianas (incluidas la mayoría de las células gramnegativas) de la muestra aparecen teñidas de verde fluorescente. Utilizando un filtro para tetrametilrodamina-5-isotiocianato, las células de C. m. subsp. sepedonicus marcadas con Cy3 aparecen teñidas de rojo fluorescente. Comparar la morfología de las células con la de los controles positivos. Las células deben ser fluorescentes brillantes y estar completamente teñidas.
Debe repetirse la prueba FISH (punto 9.4) si la tinción es aberrante. Recorrer los pocillos a lo largo de dos diámetros perpendiculares entre sí y alrededor del perímetro. En el caso de muestras que no presenten células o sólo un pequeño número de ellas es preciso observar como mínimo 40 campos microscópicos.
5.3.2 Comprobar si hay células fluorescentes brillantes con la morfología característica de C. m. subsp. sepedonicus en los pocillos de los portaobjetos (véase el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main).
La intensidad de la fluorescencia debe ser mejor que la de la cepa de control positivo o equivalente a la misma. Deberán descartarse las células que presenten una tinción incompleta o cuya fluorescencia sea escasa.
5.3.3 En el caso que se sospeche cualquier posible contaminación, deberá repetirse la prueba. Esto puede ocurrir cuando todos los portaobjetos de un lote presenten células positivas debido a la contaminación del tampón o cuando se encuentren células positivas (fuera de los pocillos) en la superficie del portaobjetos.
5.3.4 Existen varios problemas inherentes a la especificidad de la prueba FISH. En los precipitados de cuñas básales y secciones de tallo de patata pueden aparecer poblaciones de base de células fluorescentes de morfología atípica y bacterias saprofitas con reacción cruzada cuyo tamaño y morfología sean similares a los de C. m. subsp. sepedonicus, aunque con mucha menor frecuencia que en la prueba IF.
5.3.5 Deben tenerse en cuenta únicamente las células fluorescentes de tamaño y morfología típicos (véase el punto 5.3.2).
5.3.6 Interpretación de los resultados de la prueba FISH:
i) se obtendrán resultados válidos en la prueba FISH siempre que en todos los controles positivos y en ninguno de los controles negativos se observen células teñidas de verde fluorescente brillante, cuyo tamaño y morfología sean los típicos de las de C. m. subsp. sepedonicus, cuando se utilice el filtro FITC, y células teñidas de rojo fluorescente brillante cuando se utilice el filtro de rodamina. Si se encuentran células fluorescentes brillantes con morfología característica, calcular el número medio de células típicas por campo microscópico y el número de células típicas por ml de precipitado resuspendido (apéndice 4). Las muestras que presenten al menos 5 × 103 células típicas por ml de precipitado resuspendido se considerarán potencialmente contaminadas y será preciso realizar nuevas pruebas. Las muestras que presenten menos de 5 × 103 células típicas por ml de precipitado resuspendido se considerarán negativas,
ii) los resultados de la prueba FISH serán negativos cuando, utilizando el filtro de rodamina, no se observen células teñidas de rojo fluorescente brillante con un tamaño y morfología típicos de C. m. subsp. sepedonicus, siempre que se observen células teñidas de rojo fluorescente brillante típicas en las preparaciones de control positivo cuando se utilice el filtro de rodamina.
6. PRUEBA PCR.
Principio:
Cuando se utilice la prueba PCR como principal prueba de selección y ésta proporcione resultados positivos, deberá realizarse la prueba IF como segunda prueba obligatoria de selección. Cuando se utilice la prueba PCR como segunda prueba de selección y ésta proporcione resultados positivos, será preciso realizar las pruebas establecidas en el diagrama de flujo para completar el diagnóstico.
La plena explotación de este método como principal método de selección sólo se recomienda en el caso de que se hayan adquirido conocimientos especializados del mismo.
Nota: Las pruebas preliminares realizadas con este método deberán permitir la detección reproducible de 103 a 104 células de C. m. subsp. sepedonicus por ml añadidas a los extractos de muestras que previamente proporcionaron resultados negativos. Puede ser necesario llevar a cabo experimentos de optimización para lograr niveles máximos de sensibilidad y especificidad en todos los laboratorios.
Deben utilizarse reactivos y protocolos PCR validados. Seleccionar preferiblemente un método con control interno.
Tomar las precauciones necesarias para evitar la contaminación de la muestra con el ADN buscado. La prueba PCR deben llevarla a cabo técnicos experimentados en laboratorios especializados en biología molecular, con el fin de reducir al máximo la posibilidad de contaminación con el ADN buscado.
Como prueba final del procedimiento siempre deben efectuarse controles negativos (extracción de ADN y PCR) que dejen constancia de que no se ha producido arrastre de ADN.
En la prueba PCR deben incluirse los siguientes controles negativos:
‒ extracto de la muestra que previamente haya proporcionado resultados negativos para C. m. subsp. sepedonicus,
‒ controles de tampón utilizados para extraer la bacteria y el ADN de la muestra,
‒ mezcla para la reacción PCR.
Deben incluirse los siguientes controles positivos:
‒ alícuotas de los precipitados resuspendidos a los que se haya añadido C. m. subsp. sepedonicus (para la preparación, véase el apéndice 2),
‒ una suspensión de 106 células por ml de C. m. subsp. sepedonicus en agua de un aislado virulento (por ejemplo, NCPPB 2140 o NCPPB 4053),
‒ siempre que sea posible, utilizar también ADN extraído de las muestras de control positivas en la prueba PCR.
Para evitar toda posible contaminación, preparar los controles positivos en un entorno distinto al de las muestras que se vayan a someter a prueba.
Los extractos de muestras deben estar, en la medida de lo posible, libres de tierra. En determinados casos puede ser recomendable preparar las extracciones a partir de patatas lavadas cuando vayan a utilizarse protocolos PCR.
6.1 Métodos de purificación del ADN.
Utilizar muestras de control positivas y negativas, como se indica más arriba.
Procesar el material de control de la misma manera que la muestra o muestras.
Para la purificación del ADN buscado a partir de sustratos de muestras complejas, existen diversos métodos que eliminan los inhibidores de PCR y otras reacciones enzimáticas y concentran el ADN buscado en el extracto de la muestra.
El siguiente método ha sido optimizado para su utilización con el método PCR validado que figura en el apéndice 6.
6.1.a) Método de Pastrik (2000):
1. Verter con una pipeta 220 μl de tampón de lisis (100 mM NaCI, 10 mM Tris-HCI [pH 8,0], 1 mM EDTA [pH 8,0]) en un tubo eppendorf de 1,5 ml.
2. Añadir 100 μl de extracto de muestra y colocar en un calentador o al baño maría a 95 °C durante 10 minutos.
3. Colocar el tubo en hielo durante 5 minutos.
4. Añadir 80 μl de solución madre de lisozima (50 mg de lisozima por ml en 10 mM Tris HCI, pH 8,0) e incubar a 37 °C durante 30 minutos.
5. Añadir 220 μl de solución A Easy DNA® (Invitrogen), mezclar adecuadamente por agitación e incubar a 65 °C durante 30 minutos.
6. Añadir 100 μl de solución B Easy DNA® (Invitrogen) y agitar vigorosamente hasta que el precipitado se mueva libremente en el tubo y la muestra sea uniformemente viscosa.
7. Añadir 500 μl de cloroformo y agitar hasta que disminuya la viscosidad y la mezcla sea homogénea.
8. Centrifugar a 15.000 g durante 20 minutos a 4 °C para separar las fases y formar la interfase.
9. Transferir la fase superior a un tubo eppendorf sin utilizar.
10. Añadir 1 ml de etanol al 100 % (‒20 °C), agitar brevemente e incubar en hielo durante 10 minutos.
11. Centrifugar a 15.000 g durante 20 minutos a 4 °C y retirar el etanol del precipitado.
12. Añadir 500 μl de etanol al 80 % (‒20 °C) y mezclar invirtiendo el tubo.
13. Centrifugar a 15.000 g durante 10 minutos a 4 °C, guardar el precipitado y retirar el etanol.
14. Dejar que el precipitado se seque al aire o en un Speed Vac de ADN.
15. Resuspender el precipitado en 100 μl de agua ultrapura estéril y dejar a temperatura ambiente durante un mínimo de 20 minutos.
16. Almacenar a ‒20 °C hasta que se necesite para PCR.
17. Decantar todo precipitado blanco mediante centrifugación, y utilizar 5 μl del sobrenadante que contenga ADN para la prueba PCR.
6.1.b) Otros métodos:
Se pueden aplicar otros métodos de extracción de ADN (como p. ej. Qiagen DNeasy Plant Kit) siempre que hayan demostrado ser igual de eficaces para purificar el ADN de muestras de control que contengan de 103 a 104 células patógenas por ml.
6.2 PCR.
6.2.1 Preparar los ensayos controles de PCR de acuerdo con el protocolo validado (apéndice 6). Preparar una dilución decimal de extracto de ADN de la muestra (1:10 en agua ultrapura).
6.2.2 Preparar la mezcla de reacción PCR adecuada en un entorno libre de contaminación de acuerdo con el protocolo publicado (apéndice 6). El protocolo PCR validado es una reacción “multiplex” que también incorpora un protocolo PCR interno.
6.2.3 Añadir 5 μl de extracto de ADN por 25 μl de reacción PCR en tubos PCR estériles.
6.2.4 Incorporar una muestra de control negativa que contenga sólo mezcla de reacción PCR y añadir la misma fuente de agua ultrapura utilizada en la mezcla PCR en lugar de la muestra.
6.2.5 Colocar los tubos en el mismo termociclador utilizado en las pruebas preliminares y aplicar el programa PCR optimizado adecuado (apéndice 6).
6.3 Análisis del producto de la PCR.
6.3.1 Confirmar los amplicones PCR mediante electroforesis en gel de agarosa. Correr al menos 12 μl de mezcla de reacción de ADN amplificada de cada muestra mezclada con 3 μl de tampón de carga (apéndice 6) en un gel de agarosa al 2,0 % (p/v) en tampón tris-acetato-EDTA (TAE) (apéndice 6) a 5-8 V por cm. Utilizar un marcador de ADN adecuado, por ejemplo, 100 bp ladder.
6.3.2 Revelar las bandas de ADN mediante la tinción con bromuro de etidio (0,5 mg por I) durante 30 a 45 minutos, tomando las precauciones adecuadas para el manejo de este mutágeno.
6.3.3 En el caso de los productos PCR amplificados del tamaño esperado (apéndice 6), visualizar el gel teñido mediante transiluminación UV de onda corta (por ejemplo, = 302 nm) y anotar los resultados.
6.3.4 Para todos los resultados o casos nuevos, verificar la autenticidad del amplicón de PCR realizando un análisis con enzima de restricción en una muestra del ADN amplificado restante mediante la incubación a la temperatura óptima y durante el tiempo necesario con una enzima y tampón adecuados (véase el apéndice 6). Confirmar los fragmentos digeridos mediante electroforesis en gel de agarosa, siguiendo el método indicado anteriormente, y observar el patrón característico del fragmento de restricción con transiluminación UV una vez teñido con bromuro de etidio y comparar con el control positivo no digerido y digerido.
Interpretación de los resultados de la prueba PCR.
La prueba PCR es negativa si el amplicón PCR específico de la C. m. subsp. sepedonicus del tamaño esperado no se detecta en la muestra en cuestión pero sí en todas las muestras de los controles positivos (en el caso de PCR “multiplex” con cebadores de control interno específicos de la planta, se debe amplificar con la muestra en cuestión un segundo producto PCR del tamaño esperado).
La prueba PCR es positiva si se detecta el amplicón PCR específico de la C. m. subsp. sepedonicus del tamaño y patrón de restricción (cuando sea necesario) esperados, siempre que no se amplifique a partir de ninguna de las muestras de los controles negativos. La confirmación fiable de un resultado positivo puede obtenerse también repitiendo la prueba con un segundo conjunto de cebadores PCR (punto 9.3).
Nota: Se puede sospechar la inhibición de la PCR si el amplicón esperado se obtiene de la muestra de control positiva que contiene C. m. subsp. sepedonicus en agua pero se obtienen resultados negativos de los controles positivos con C. m. subsp. sepedonicus en extracto de patata. En protocolos PCR “multiplex” con controles PCR internos, la inhibición de la reacción aparece indicada cuando no se obtiene ninguno de los dos amplicones.
Se puede sospechar la existencia de contaminación si el amplicón esperado se obtiene a partir de uno o varios de los controles negativos.
7. PRUEBA DE BIOENSAYO.
Nota: Las pruebas preliminares realizadas con este método deberán permitir la detección reproducible de 103 a 104 unidades de formación de colonias de C. m. subsp. sepedonicus por ml añadidas a los extractos de muestras que previamente proporcionaron resultados negativos (para la preparación véase el apéndice 2).
La máxima sensibilidad de detección se alcanzará cuando se utilicen extractos de muestras que se acaben de preparar y cuando las condiciones de cultivo sean las óptimas. No obstante, este método puede aplicarse también con éxito a extractos que se hayan almacenado en glicerol a una temperatura comprendida entre ‒68 y ‒86 °C.
Algunas variedades de berenjena constituyen un medio de enriquecimiento selectivo excelente para la proliferación de C. m. subsp. sepedonicus, incluso en ausencia de síntomas, y facilitan también una prueba confirmatoria de huésped excelente.
Las condiciones de cultivo deben ser óptimas para reducir el riesgo de obtener falsos resultados negativos en las pruebas.
Para más detalles sobre el cultivo, véase el apéndice 8.
7.1 Distribuir la totalidad de la alícuota de prueba sobrante del precipitado resuspendido de los puntos 3.1.6 o 3.2.5 entre las berenjenas, siguiendo uno de los métodos que se exponen a continuación (7.3 o 7.4). Utilizar exclusivamente plantas que se encuentren en la fase foliar 2 - 3 y hasta la plena expansión de la tercera hoja verdadera. A fin de garantizar la utilización completa del precipitado resuspendido, así como la inoculación efectiva, se requerirán entre 15 y 25 berenjenas por muestra para los procedimientos que se esbozan a continuación.
7.2 Las berenjenas no deben regarse de uno a dos días antes de la inoculación para reducir la presión de turgencia.
7.3 Inoculación en estrías.
7.3.1 Sosteniendo la planta entre dos dedos aplíquese con una pipeta una gota (de unos 5-10 μl) del precipitado en suspensión en el tallo situado entre los cotiledones y la primera hoja.
7.3.2 Con un bisturí estéril practicar una incisión diagonal de aproximadamente 1,0 cm de largo y una profundidad de aproximadamente 2/3 del grosor del tallo, comenzando el corte a partir de la gota del precipitado.
7.3.3 Sellar el corte con vaselina estéril con una jeringa.
7.4 Inoculación con jeringa.
Inocúlense los tallos de la berenjena justo por encima de los cotiledones utilizando una jeringa provista de aguja hipodérmica (no menos de 23G). Distribuir la muestra entre las berenjenas.
7.5 Como controles positivos, inocular 5 plantas con una suspensión acuosa de 105 a 106 células por ml de un cultivo conocido de C. m. subsp. sepedonicus y, siempre que sea posible, con tejido de tubérculo infectado de forma natural (véase el punto 4) mediante el mismo método de inoculación (7.3 o 7.4).
7.6 Como control negativo, inocular 5 plantas con tampón de precipitado estéril mediante el mismo método de inoculación (7.3 o 7.4).
7.7 Incubar las plantas en instalaciones adecuadas para la cuarentena durante un máximo de 4 semanas a una temperatura de 18 a 24 °C. Incubar las plantas con suficiente luz y un elevado nivel de humedad (70 a 80 %) y agua, evitando que el agua se estanque y que las plantas se marchiten por falta de agua. Las células de C. m. subsp sepedonicus mueren a temperaturas superiores a 30 °C y la temperatura óptima es 21 °C. Para evitar la contaminación se deben incubar las plantas de control positivo y negativo en bancos claramente separados en un invernadero o cámara de cultivo; en el caso de que se disponga de un espacio reducido, garantizar la estricta separación entre tratamientos. En el caso de que plantas para distintas muestras deban incubarse juntas, deben separarse con las pantallas adecuadas. Durante la fertilización, el riego, la inspección y cualquier otra manipulación deben extremarse las precauciones para evitar la contaminación cruzada. Es esencial mantener los invernaderos y las cámaras libres de toda plaga de insectos, ya que estos pueden transmitir la bacteria de una muestra a otra.
7.8 Transcurrida una semana, examinar con regularidad a fin de detectar los síntomas. Contar el número de plantas que muestren síntomas. C. m. subsp. sepedonicus provoca el marchitamiento de las hojas en las berenjenas, que puede iniciarse con flacidez internervial o de los bordes. El tejido marchito puede aparecer al principio de color verde oscuro o moteado, pero se vuelve más pálido antes de necrosarse. Las zonas de marchitamiento internerviales suelen tener un aspecto graso o húmedo como empapados en agua. El tejido necrosado presenta a veces un borde amarillo brillante. Las plantas no están necesariamente muertas; cuanto más tardan en aparecer los síntomas, tanto mayor es la posibilidad de supervivencia. Las plantas pueden superar la infección. Las berenjenas jóvenes son mucho más sensibles a las poblaciones bajas de C. m. subsp. sepedonicus que las plantas de más edad, por lo que es necesario utilizar plantas en el estadio foliar 3, o inmediatamente antes del mismo.
El marchitamiento puede estar inducido también por poblaciones de otras bacterias u hongos presentes en el precipitado de tejido tuberoso. Entre estos se incluyen Ralstonia solanacearum, Erwinia carotovora subsp. carotovora y E. carotovora subsp. atroseptica, Erwinia chrysanthemi, Phoma exigua var. foveata, así como grandes poblaciones de bacterias saprofitas. En particular, Erwinia chrysanthemi puede provocar síntomas y marchitamiento en las hojas muy parecidos a los de C. m. subsp sepedonicus. La única diferencia en el caso de las infecciones causadas por Erwinia chrysanthemi es el ennegrecimiento de los tallos. Dichos marchitamientos se pueden distinguir de los causados por C. m. subsp. sepedonicus, porque se marchitan rápidamente hojas o plantas enteras. También puede prepararse una tinción de Gram, esta prueba diferenciará a C. m. subsp. sepedonicus de Erwinia ssp.
7.9 Tan pronto como se observen síntomas en las berenjenas deberá procederse al reaislamiento, utilizando secciones de tejido foliar o del tallo de las plantas marchitas (véase el punto 3.1.3 para la maceración del tejido). Desinfectar la superficie de las hojas y tallos de berenjena frotándolos con etanol al 70 %. Realizar una prueba IF o PCR con la savia de la planta de berenjena y aislar en medios (selectivos) adecuados (véase el punto 8). También puede prepararse una tinción de Gram (apéndice 9). Identificar los cultivos purificados de presunta C. m. subsp. sepedonicus y confirmar la patogenicidad (véanse las secciones 9 y 10).
7.10 En determinadas circunstancias, en particular cuando las condiciones de cultivo no sean óptimas, puede ocurrir que C. m. subsp. sepedonicus esté presente de forma latente en las berenjenas, incluso después de períodos de incubación de hasta 4 semanas. En el caso de que, transcurridas 4 semanas, no se observen síntomas, realizar una prueba IF/PCR con una muestra mixta de secciones de tallo de 1 cm de cada planta tomadas por encima de la sección de inoculación. Si los resultados de la prueba son positivos, deberá procederse al reaislamiento en medios (selectivos) adecuados de acuerdo con el procedimiento descrito en el punto 8. Identificar los cultivos purificados de posible C. m. subsp. sepedonicus y confirmar la patogenicidad (secciones 9 y 10).
Interpretación de los resultados de la prueba del bioensayo.
Se obtendrán resultados válidos en la prueba del bioensayo cuando las plantas de control positivo muestren síntomas típicos, se puedan reaislar las bacterias de dichas plantas y no se detecten síntomas en los controles negativos.
Los resultados de la prueba del bioensayo serán negativos si las plantas sometidas a la prueba no están infectadas por C. m. subsp. sepedonicus y siempre que se detecte la C. m. subsp. sepedonicus en los controles positivos.
Los resultados de la prueba del bioensayo serán positivos cuando las plantas sometidas a la prueba estén infectadas por C. m. subsp. sepedonicus.
8. AISLAMIENTO DE C. m. subsp. sepedonicus.
Nota: El diagnóstico sólo estará completo cuando se haya aislado e identificado (véase el punto 9) C. m. subsp. sepedonicus, y posteriormente confirmado mediante una prueba de patogenicidad (punto 10). Aunque C. m. subsp sepedonicus es un organismo delicado, puede aislarse a partir de tejido sintomático.
No obstante, puede verse inhibida por las bacterias saprofitas que proliferan rápidamente y, por tanto, los aislamientos directos a partir del precipitado de tejido tuberoso o del tallo (punto 3.1.6 o 3.2.5) son difíciles. El aislamiento directo de C. m. subsp. sepedonicus se puede realizar con un medio selectivo y la dilución adecuada del precipitado resuspendido de las cuñas básales o tallos de patatas.
Los aislamientos se realizarán con todos los tubérculos o secciones de tallo de patata sintomáticos y con las berenjenas en las no se observen síntomas pero con las que la prueba IF/PCR realizada a partir de la muestra mixta haya arrojado resultados positivos (véase el punto 7.10). Cuando sea necesario, la maceración de los tallos de berenjena deberá realizarse siguiendo el procedimiento descrito en el punto 3.1.3.
Como controles positivos preparar diluciones decimales a partir de una suspensión de 106 cfu por ml de C. m. subsp. sepedonicus (p. ej. NCPPB 4053 o PD 406). Para evitar cualquier posibilidad de contaminación, preparar los controles positivos separadamente de las muestras que se vayan a someter a prueba.
Cada lote de medio selectivo que se prepare deberá someterse a prueba a fin de determinar su idoneidad para el cultivo del patógeno con anterioridad a su utilización en pruebas con muestras rutinarias.
Procesar el material de control de la misma manera que la muestra o muestras.
8.1 Siembra en medio selectivo.
8.1.1 A partir de una alícuota de 100 μl de una muestra de precipitado de patata resuspendido o de savia de berenjena, realizar diluciones decimales en tampón de precipitado (apéndice 3).
8.1.2 El aislamiento a partir de precipitado de patata sin diluir no suele funcionar debido a la lentitud de crecimiento de C. m. subsp sepedonicus y a la competencia de saprofitas. Dado que en los tejidos infectados suele haber poblaciones elevadas de la bacteria, las saprofitas se diluyen normalmente, mientras que el patógeno persiste. Se recomienda, por tanto, recurrir a la técnica de la extensión en placa y extender, con ayuda de un asa de extensión (“palos de hockey”), 100 μl de cada una de las muestras, en diluciones de 1/100 hasta 1/10 000, en medio MTNA o NCP-88 (apéndice 5) (si se utilizan placas de Petri de 90 mm de diámetro, se deberá ajustar el volumen para tamaños alternativos de placa).
Nota: Otra estrategia alternativa consiste en extender la alícuota inicial de 100 μl de precipitado de patata en una primera placa de agar con ayuda de un aplicador y posteriormente extender, en estrías, todos los residuos que queden en el aplicador en una segunda placa de agar. Repetir por último con una tercera placa, logrando así, mediante el aplicador, el efecto de dilución en las placas.
8.1.3 Incubar las placas en la oscuridad a una temperatura de 21 a 23 °C.
8.1.4 Las observaciones iniciales de las placas, incluidos los recuentos de colonias típicas de la C. m. subsp. sepedonicus en relación con las placas de control, se realizan 3 días después, con recuentos posteriores transcurridos 5, 7 y, finalmente, 10 días.
8.2 Purificación de las colonias sospechosas.
Nota: El subcultivo de colonias de aspecto similar a C. m. subsp. sepedonicus debe realizarse en medios YMG para la inoculación de berenjenas y/o la posterior identificación. Esto debe realizarse antes de que las placas crezcan demasiado, es decir, preferiblemente transcurridos de 3 a 5 días.
8.2.1 Practicar una siembra en estrías de colonias de aspecto similar a la C. m. subsp. sepedonicus en la superficie de uno de los siguientes medios (apéndice 5): agar de dextrosa nutritivo (NAD) (para su utilización exclusiva en el subcultivo), agar de levadura, peptona, glucosa (YPGA), agar de extracto de levadura, sales minerales (YGM).
Incubar a 21-24 °C durante 10 días como máximo.
C. m. subsp. sepedonicus crece lentamente y suele provocar unas colonias punteadas, de color crema, de forma abovedada, en el plazo de diez días. [Fotos de colonias típicas de C. m. subsp. sepedonicus (véase el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main)].
8.2.2 Volver a sembrar en estrías para obtener cultivos puros.
El ritmo de crecimiento mejora con subcultivos. Las colonias típicas son de color blanco crema o marfil, amarillas en ocasiones, redondeadas, lisas, abultadas, abovedadas en forma convexa, mucoso-fluidas, con bordes intactos y normalmente de un diámetro de 1 a 3 mm.
Una simple tinción de Gram (apéndice 9) puede resultar útil para seleccionar colonias para pruebas posteriores.
8.2.3 Identificar los supuestos cultivos (véase el punto 9) y realizar una prueba de patogenicidad (véase el punto 10).
9. IDENTIFICACIÓN.
Identificar cultivos puros de posibles aislados de C. m. subsp. Sepedonicus, utilizando al menos dos de las pruebas siguientes basadas en principios biológicos diferentes.
Cuando proceda, incluir cepas de referencia conocidas para cada prueba efectuada, como controles positivos.
9.1 Pruebas de identificación nutricional y enzimática.
Determinar las propiedades fenotípicas siguientes que estén universalmente presentes o ausentes en la C. m. subsp. sepedonicus, de acuerdo con los métodos de Lelliott y Stead (1987), Klement et al. (1990), Schaad (2001), Anónimo (1987).
Incubar todos los medios a 21 °C y examinar transcurridos 6 días. Si no se ha producido crecimiento, incubar 20 días.
En todas las pruebas deberá incluirse un control conocido de C. m. subsp. sepedonicus. Las pruebas fisiológicas y nutricionales deberán realizarse utilizando inoculaciones de subcultivos en agar nutritivo. Las comparaciones morfológicas deberán realizarse a partir de cultivos de agar de dextrosa nutritivos.
| Pruebas | Resultado esperado |
|---|---|
| Prueba de la oxidación/fermentac. (O/F) | Inerte o ligeramente oxidante |
| Actividad oxidasa | ‒ |
| Crecimiento a 37 °C | ‒ |
| Actividad ureasa | ‒ |
| Hidrólisis de la esculina | + |
| Hidrólisis del almidón | ‒ o débil |
| Tolerancia de NaCI al 7 % | ‒ |
| Producción de indol | ‒ |
| Actividad catalasa | + |
| Producción de H2S | ‒ |
| Utilización de citrato | ‒ |
| Licuefacción de la gelatina | ‒ |
| Producción de ácido de glicerol | ‒ |
| Producción de ácido de lactosa | ‒ o débil |
| Producción de ácido de ramnosa | ‒ |
| Producción de ácido de salicina | ‒ |
| Tinción de Gram (apéndice 9) | + |
9.2 Prueba IF.
a) Preparar una suspensión de aproximadamente 106 células por ml en tampón IF (apéndice 3).
b) Preparar una serie de diluciones a 1/2 de un antisuero adecuado.
c) Aplicar el procedimiento IF (punto 4).
d) La prueba IF será positiva si el título IF del cultivo es equivalente al del control positivo.
9.3 Prueba PCR.
a) Preparar una suspensión de aproximadamente 106 células por ml en agua ultrapura.
b) Calentar 100 μl de la suspensión celular en tubos cerrados en un calentador o al baño maría a 100 °C durante 4 minutos. Si fuese necesario, la adición de NaOH recién preparado a una concentración final de 0,05 M puede ayudar a la lisis celular. Las muestras pueden almacenarse entonces a una temperatura de ‒16 a ‒24 °C hasta que sea necesario.
c) Aplicar los procedimientos PCR adecuados para amplificar los amplicones específicos de C. m. subsp. sepedonicus (por ejemplo, Pastrik, 2000; véase el apéndice 4; Li y de Boer, 1995; Mills et al., 1997; Pastrik y Rainey, 1999; Schaad et al., 1999).
d) Se logrará la identificación positiva de C. m. subsp. sepedonicus si los amplicones de PCR son del mismo tamaño y presentan polimorfismos del fragmento de restricción con la misma longitud que los de la cepa de control positivo.
9.4 Prueba FISH.
a) Preparar una suspensión de aproximadamente 106 células por ml en agua ultrapura.
b) Aplicar el procedimiento FISH (punto 5).
c) La prueba FISH será positiva si se obtienen las mismas reacciones del cultivo y el control positivo.
9.5 Perfiles de ácidos grasos (Fatty acid profiling o FAP).
a) Mantener el cultivo en agar de tripticasa de soja (Oxoid) durante 72 horas a 21 °C (+/‒1o).
b) Aplicar un procedimiento FAP adecuado (Janse, 1991; Stead, 1992).
c) La prueba FAP será positiva si el perfil del supuesto cultivo es idéntico al del control positivo. Los ácidos grasos cuya presencia es característica son 15:1 Anteiso A, 15:0 Iso, 15:0 Anteiso, 16:0 Iso, 16:0 y 17:0. El Anteiso es altamente indicativo de C. m. subsp sepedonicus. Otros géneros tales como Curtobacterium, Arthrobacter y Micrococcus cuentan también con algunos de estos ácidos. No obstante, el 15:1 Anteiso A es un ácido raro en estas bacterias que, sin embargo, está presente en todas las spp. Clavibacter en una proporción que oscila entre el 1 y el 5 %. En C. m. subsp sepedonicus el valor se sitúa normalmente en torno al 5 %.
9.6 BOX-PCR.
a) Preparar una suspensión de aproximadamente 106 células por ml en agua ultrapura.
b) Aplicar la prueba con arreglo al procedimiento (Smith et al., 2001).
10 PRUEBA DE CONFIRMACIÓN DE PATOGENCIDAD.
Como confirmación final del diagnóstico de C. m. subsp. sepedonicus y para la evaluación de la virulencia de cultivos identificados como C. m. subsp. sepedonicus debe realizarse la prueba de patogenicidad.
10.1 Preparar un inoculo de aproximadamente 106 células por ml de cultivos de 3 días del aislado que se vaya a someter a prueba y de una cepa de control positivo adecuada de C. m. subsp. sepedonicus.
10.2 Inocular entre 5 y 10 tallos de plántulas de berenjena en la fase de la tercera hoja (punto 7.3 o 7.4).
10.3 Incubar a una temperatura de 18 a 24 °C, con luz suficiente, humedad relativa alta y riego adecuado, evitando tanto el estancamiento del agua como el estrés provocado por la sequía (punto 7.7) Con los cultivos puros deberá obtenerse el marchitamiento típico en el plazo de dos semanas.
Transcurrido este plazo, las plantas que no muestren síntoma alguno (véase el punto 7.8) deberán incubarse durante un máximo de 3 semanas a temperaturas que favorezcan su crecimiento pero que no superen los 25 °C (apéndice 8). Si después de 3 semanas no se presentan los síntomas, no podrá confirmarse que el cultivo es una forma patógena de C. m. subsp. sepedonicus.
10.4 Aislar de las plantas sintomáticas separando una sección de tallo que esté 2 cm por encima de la sección de inoculación. Dilacerar y suspender en un pequeño volumen de agua destilada estéril o en tampón fosfato 50 mM (apéndice 3). Aislar de la suspensión mediante dilución, extendiendo o aplicando en estrías en MTNA e YPGA (apéndice 5), incubar entre 3 y 5 días a una temperatura de 21 a 23 °C y examinar la formación de colonias típicas de C. m. subsp. sepedonicus.
APÉNDICE 1
Laboratorios dedicados a la optimización y la validación de los protocolos
| Laboratorio (1) | Ciudad | País |
|---|---|---|
| Agentur für Gesundheit und Ernährungssicherheit | Viena y Linz | Austria |
| Departement Gewasbescherming | Merelbeke | Bélgica |
| Plantedirektoratet | Lyngby | Dinamarca |
| Central Science Laboratory | York | Inglaterra |
| Scottish Agricultural Science Agency | Edimburgo | Escocia |
| Laboratoire National de la Protection des Végétaux, Unité Bactériologie | Angers | Francia |
| Laboratoire National de la Protection des Végétaux, Station de Quarantaine de la Pomme de Terre | Le Rheu | Francia |
| Biologische Bundesanstalt | Kleinmachnow | Alemania |
| Pflanzenschutzamt Hannover | Hannover | Alemania |
| State Laboratory | Irlanda | Dublín |
| Plantenziektenkundige Dienst | Wageningen | Países Bajos |
| Norwegian Crop Research Institute, Plant Protection Centre | Aas | Noruega |
| Direcção-Geral de Protecção das Culturas | Lisboa | Portugal |
| Nacionalni institut za biologijo | Liubliana | Eslovenia |
| Centro de Diagnóstico de Aldearrubia | Salamanca | España |
(1) Científicos de contacto: véase el sitio web http://forum.europa.eu.int/Public/irc/sanco/Home/main.
APÉNDICE 2
Preparación de controles positivos y negativos para las pruebas de selección básicas PCR/IF y FISH
Preparar un cultivo de 72 horas de una cepa virulenta de C. m. subsp. sepedonicus [NCPPB 4053 o PD 406] en medio base MTNA y suspender en tampón fosfato 10 mM para obtener una concentración de aproximadamente 1 a 2 × 108 cfu por ml. Esto se obtiene normalmente mediante una suspensión ligeramente turbia equivalente a una densidad óptica de 0,20 a 600 nm.
Separar las cuñas básales de 200 tubérculos de una variedad de piel blanca conocida por estar exenta de C. m. subsp. sepedonicus.
Procesar las cuñas básales como siempre y resuspender el precipitado en 10 ml.
Preparar 10 microviales estériles de 1,5 ml con 900 μl del precipitado resuspendido.
Transferir 100 μl de la suspensión de C. m. subsp. sepedonicus al primer microvial. Homogeneizar por agitación.
Establecer niveles decimales de contaminación realizando nuevas diluciones en los 5 microviales siguientes.
Los 6 microviales contaminados se utilizarán como controles positivos. Los 4 microviales no contaminados se utilizarán como controles negativos. Etiquetar los microviales como corresponda.
Preparar alícuotas de 100 μl en microviales estériles de 1,5 ml para obtener de este modo 9 réplicas de cada muestra de control. Almacenar a una temperatura de ‒16 a ‒24 °C hasta su uso.
La presencia y la cuantificación de C. m. subsp. sepedonicus en las muestras de control deberá confirmarse en primer lugar mediante una prueba IF.
Para la prueba PCR, extraer ADN de las muestras de control positivas y negativas para cada serie de muestras de ensayo.
Para las pruebas IF y FISH, realizar ensayos con las muestras de control positivas y negativas para cada serie de muestras de ensayo.
En las pruebas IF, FISH y PCR, debe detectarse C. m. subsp. sepedonicus en al menos 106 y 104 células/ml de los controles positivos y en ninguno de los controles negativos.
APÉNDICE 3
Tampones para los métodos de prueba
GENERAL: Los tampones esterilizados pueden almacenarse sin abrir hasta un año.
1. Tampones para el procedimiento de extracción
1.1 Tampón de extracción (tampón fosfato 50 mM, pH 7,0)
Este tampón se utiliza para la extracción de la bacteria del tejido de las plantas mediante homogeneización o agitación.
Na2HPO4 (anhidro): 4,26 g
KH2PO4: 2,72 g
Agua destilada: 1,00 I
Disolver los ingredientes, verificar el pH y esterilizar en autoclave a 121 °C durante 15 minutos.
Los siguientes componentes adicionales pueden resultar útiles:
| Finalidad | Cantidad (por I) | |
|---|---|---|
| Copos de Lubrol | Defloculante (*) | 0,5 g |
| Compuesto DC silicona antiespumante | Antiespumante (*) | 1,0 ml |
| Pirofosfato tetrasódico | Antioxidante | 1,0 g |
| Polivinilpirrolidona -40 000 (PVP-40) | Unir los inhibidores PCR | 50 g |
(*) Para su utilización en el método de extracción por homogeneización.
1.2 Tampón de extracción (tampón fosfato 10 mM, pH 7,2)
Este tampón se utiliza para la resuspensión y la dilución de extractos de cuñas básales de tubérculos de patata tras la concentración en un precipitado mediante centrifugación.
Na2HPO4.12H2O: 2,7 g
NaH2PO4.2H2O: 0,4 g
Agua destilada: 1,00 l
Disolver los ingredientes, verificar el pH y esterilizar en autoclave a 121 °C durante 15 minutos.
2. Tampones para la prueba IF.
2.1 Tampón IF [tampón fosfato salino (PBS) 10 mM, pH 7,2].
Este tampón se utiliza para la difusión de anticuerpos
Na2HPO4.12H2O: 2,7 g
NaH2PO4.2H2O: 0,4 g
NaCl: 8,0 g
Agua destilada: 1,00 l
Disolver los ingredientes, verificar el pH y esterilizar en autoclave a 121 °C durante 15 minutos.
2.2 Tampón IF-Tween.
Este tampón se utiliza para lavar los portaobjetos.
Añadir Tween 20 al 0,1 % al tampón IF.
2.3 Solución de glicerol con tampón fosfato, pH 7,6.
Este tampón se utiliza como fluido de montaje en los pocillos de los portaobjetos de IF para aumentar la fluorescencia.
Na2HPO4.12H2O: 3,2 g
NaH2PO4.2H2O: 0,15 g
Glicerol: 50 ml
Agua destilada: 100 ml
En el mercado se pueden encontrar soluciones de montaje que protegen la fluorescencia, como p. ej., Vectashield® (Vector Laboratories) o Citifluor® (Leica).
APÉNDICE 4
Determinación del nivel de contaminación en las pruebas IF y FISH
1. Contar el número de células fluorescentes típicas por campo de visión (c).
2. Calcular el número de células fluorescentes típicas por pocillo del portaobjetos del microscopio (C).
C = c × S/s
donde:
S = superficie del pocillo del portaobjetos múltiple y
s = superficie del campo del objetivo
s = πi2/4G2K2
donde:
i = coeficiente de campo (varía de 8 a 24 dependiendo del tipo ocular)
K = coeficiente del tubo (1 o 1,25)
G = aumentos del objetivo (100x, 40x, etc.)
3. Calcular el número de células fluorescentes típicas por ml de precipitado resuspendido (N).
N = C × 1.000/y × F
donde:
y = volumen de precipitado resuspendido en cada pocillo y
F = factor de dilución del precipitado resuspendido.
APÉNDICE 5
Medios para el aislamiento y cultivo de C. m. subsp. sepedonicus
a) Medios de cultivo generales:
Agar nutritivo (NA)
Agar nutritivo (Difco): 23,0 g
Agua destilada: 1,00 I
Disolver los ingredientes y esterilizar en autoclave a 121 °C durante 15 minutos.
Agar de dextrosa nutritivo (NDA)
Agar nutritivo de Difco Bacto con un 1 % de D(+)-glucosa (monohidrato). Esterilizar en autoclave a 115 °C durante 20 minutos.
Agar de levadura, peptona, glucosa (YPGA)
Extracto de levadura (Difco): 5,0 g
Bacto-peptona (Difco): 5,0 g
D(+)-glucosa (monohidrato): 10,0 g
Bacto-agar (Difco): 15,0 g
Agua destilada: 1,00 I
Disolver los ingredientes y esterilizar en autoclave a 121 °C durante 15 minutos.
Medio de sales minerales, extracto de levadura (YGM)
Bacto-extracto de levadura (Difco): 2,0 g
D(+)-glucosa (monohidrato): 2,5 g
K2HPO4: 0,25 g
KH2PO4: 0,25 g
MgSO4.7H2O: 0,1 g
MnSO4.H2O: 0,015 g
NaCI: 0,05 g
FeSO4.7H2O: 0,005 g
Bacto-agar (Difco): 18 g
Agua destilada: 1,00 I
Disolver los ingredientes y esterilizar volúmenes de medio litro de este medio en autoclave a 115 °C durante 20 minutos.
b) Medios de cultivo selectivo validados:
Medio MTNA:
A menos que se especifique de otra manera, todos los componentes de los medios son de BDH.
Extracto de levadura (Difco): 2,0 g
Manitol: 2,5 g
K2HPO4: 0,25 g
KH2PO4: 0,25 g
NaCI: 0,05 g
MgSO4.7H2O: 0,1 g
MnSO4.H2O: 0,015 g
FeSO4.7H2O: 0,005 g
Agar (Oxoid no 1): 16,0 g
Agua destilada: 1,0 I
Disolver los ingredientes y ajustar el pH a 7,2. Tras esterilizar en autoclave (a 121 °C durante 15 minutos) y enfriar a 50 °C, añadir los siguientes antibióticos: 0,06 g de trimetoprim, 0,002 g de ácido nalidíxico y 0,01 g de anfotericina B.
Soluciones antibióticas estándar: trimetoprim (Sigma) y ácido nalidíxico (Sigma) (en ambos casos a 5 mg/ml), en metanol al 96 %, anfotericina B (Sigma) (1 mg/ml) en dimetil sulfóxido. Las soluciones estándar se esterilizan por filtración.
Nota: La durabilidad del medio base es de 3 meses. Una vez añadidos los antibióticos, la durabilidad es de 1 mes siempre que se mantengan refrigerados.
Medio NCP-88
Agar nutritivo (Difco): 23 g
Extracto de levadura (Difco): 2 g
D-manitol: 5 g
K2HPO4: 2 g
KH2PO4: 0,5 g
MgSO4.7H2O: 0,25 g
Agua destilada: 1,0 I
Disolver los ingredientes y ajustar el pH a 7,2. Tras esterilizar en autoclave y enfriar a 50 °C, añadir los siguientes antibióticos: 0,003 g de sulfato de polimixina B (Sigma), 0,008 g de ácido nalidíxico (Sigma) y 0,2 g de cicloheximida (Sigma).
Disolver los antibióticos en las siguientes soluciones estándar: el ácido nalidíxico en NaOH 0,01 M, la cicloheximida en etanol al 50 % y el sulfato de polimixina B en agua destilada. Las soluciones estándar se esterilizan por filtración.
Nota: La durabilidad del medio base es de 3 meses. Una vez añadidos los antibióticos, la durabilidad es de 1 mes siempre que se mantengan refrigerados.
APÉNDICE 6
Reactivos y protocolo PCR validados
Nota: Las pruebas preliminares deberán permitir la detección reproducible de al menos 103 a 104 células de C. m. subsp sepedonicus por ml de extracto de muestra.
Cebador directo PSA-1
Cebador reverso PSA -R
Cebador directo NS-7-F
Cebador reverso NS-8-R
Por otra parte, las pruebas preliminares no deben proporcionar resultados falsos positivos con respecto a un conjunto de cepas bacterianas seleccionadas.
1. Protocolo PCR “multiplex” con control PCR interno (Pastrik, 2000)
1.1 Cebadores oligonucleótidos
5'- ctc ctt gtg ggg tgg gaa aa -3'
5'- tac tga gat gtt tea ctt ccc c -3'
5'- gag gca ata aca ggt ctg tga tgc -3'
5'- tcc gca ggt tea cct acg ga -3'
Tamaño esperado del amplicón de ADN de C. m. subsp. sepedonicus = 502 pb (conjunto de cebadores PSA).
Tamaño esperado del amplicón del control PCR interno de ARNr 18S = 377 pb (conjunto de cebadores NS).
1.2 Mezcla para la reacción PCR.
| Reactivo | Cantidad para la reacción | Concentración final |
|---|---|---|
| Agua ultrapura estéril | 15,725 μl | |
| 10x tampón PCR (1) (15 mM MgCl2) | 2,5 μl | 1x (1,5 mM MgCl2) |
| BSA (fracción V) (10 %) | 0,25 μl | 0,1 % |
| Mezcla d-nTP (20 mM) | 0,125 μl | 0,1 mM |
| Cebador PSA-1 (10pM) | 0,5 μl | 0,2 μM |
| Cebador PSA-R (10pM) | 0,5 μl | 0,2 μM |
| Cebador NS-7-F (10pM) (2) | 0,1 μl | 0,04 μM |
| Cebador NS-8-R (10pM) (2) | 0,1 μl | 0,04 μM |
| Polimerasa Taq (5U/pl) (1) | 0,2 μl | 1,0 U |
| Volumen de muestra | 5,0 μl | |
| Volumen total: | 25,0 μl |
(1) Los métodos se validaron utilizando polimerasa Taq de Perkin Elmer (AmpliTaq o Gold) y Gibco BRL.
(2) La concentración de los cebadores NS-7 F y NS-8-R se optimizó para la extracción de cuñas básales de patata utilizando el método de homogeneización y purificación del ADN de acuerdo con Pastrik (2000) (véanse los puntos 6.1.a) y 6.2). Será necesario proceder a la reoptimización de las concentraciones de reactivos si se utiliza el método de extracción por agitación u otros métodos de aislamiento del ADN.
1.3 Condiciones de reacción PCR.
Aplicar el siguiente programa:
1 ciclo de:
i) 3 minutos a 95 °C (desnaturalización de ADN)
10 ciclos de:
ii) 1 minuto a 95 °C (desnaturalización de ADN)
iii) 1 minuto a 64 °C (anillamiento de los cebadores)
iv) 1 minuto a 72 °C (extensión de la copia)
25 ciclos de:
v) 30 segundos a 95 °C (desnaturalización de ADN)
vi) 30 segundos a 62 °C (anillamiento de los cebadores)
vii) 1 minuto a 72 °C (extensión de la copia)
1 ciclo de:
viii) 5 minutos a 72 °C (extensión final)
ix) mantener a 4 °C
Nota: Este programa se ha optimizado para utilizarlo con un termociclador MJ Research PTC 200. Puede ser preciso modificar la duración de los ciclos ii), iii), iv), v), vi) y vii) para su utilización con otros modelos.
1.4 Análisis de la enzima de restricción del amplicón.
Los productos PCR amplificados a partir de ADN de C. m. subsp. sepedonicus producen un polimorfismo de la longitud del fragmento de restricción característico con la enzima Bgl II tras la incubación a 37 °C durante 30 minutos. Los tamaños de los fragmentos de restricción obtenidos a partir del fragmento específico de C. m. subsp. sepedonicus son de 282 pb y 220 pb.
2. Preparación del tampón de carga:
2.1 Azul de bromofenol (solución estándar al 10 %):
Azul de bromofenol: 5 g
Agua bidestilada: 50 ml
2.2 Tampón de carga:
Glicerol (86 %): 3,5 ml
Azul de bromofenol (2.1): 300 μl
Agua bidestilada: 6,2 ml
3. 10X tampón de tris acetato EDTA (TAE), pH 8,0:
Tampón tris: 48,4 g
Ácido acético glacial: 11,42 ml
EDTA (sal disódica): 3,72 g
Agua destilada: 1,0 l
Diluir a 1X antes de utilizarlo.
Se encuentra también disponible en el mercado (por ejemplo, Invitrogen o equivalente).
APÉNDICE 7
Reactivos validados para la prueba FISH
1. Oligosondas.
Sonda específica para Cms CMS-CY3-01:
5'-ttg cgg ggc gca cat ctc tgc acg -3'
Sonda eubacteriana no específica EUB-338-FITC:
5'-gct gcc tcc cgt agg agt-3'
2. Solución fijadora.
[ADVERTENCIA: LA SOLUCIÓN FIJADORA CONTIENE PARAFORMALDEHÍDO QUE ES TÓXICO. UTILIZAR GUANTES Y NO INHALAR. SE RECOMIENDA TRABAJAR EN UNA CAMPANA EXTRACTORA DE GASES.]
i) Calentar 9 ml de agua de grado molecular (p. ej. agua ultrapura) a 60 °C aproximadamente y añadir 0,4 g de paraformaldehído. El paraformaldehído se disuelve cuando se añaden 5 gotas de NaOH 1N y se remueve con un agitador magnético.
ii) Ajustar el pH a 7,0 mediante la adición de 1 ml de tampón fosfato de concentración 0,1 M (pH 7,0) y 5 gotas de HCI 1N. Verificar el pH con bandas indicadoras y ajustar si fuese necesario con HCI o NaOH.
[ADVERTENCIA: NO UTILIZAR UN MEDIDOR DE PH EN SOLUCIONES QUE CONTENGAN PARAFORMALDEHÍDO.]
iii) Filtrar la solución con un filtro de membrana de 0,22 pm y mantener libre de polvo a 4 °C hasta nueva utilización.
iv) Nota: Solución fijadora alternativa: etanol al 96 %.
3. 3X Hybmix.
NaCI: 2,7 M
Tris-HCI: 60 mM (pH 7,4)
EDTA (esterilizado por filtración y autoclave): 15 mM
Diluir a 1X cuando se precise.
4. Solución de hibridación.
1X Hybmix:
Dodecilsulfato sódico (SDS): 0,01 %
sonda EUB 338: 5 ng/μl
sonda CMSCY301: 5 ng/μl
Preparar las cantidades de solución de hibridación de acuerdo con los cálculos de la tabla 1. Para cada portaobjetos (que contenga 2 muestras distintas por duplicado) se precisan 90 μl de solución de hibridación.
Tabla 1. Cantidades sugeridas para la preparación de la mezcla de hibridación
| 2 portaobjetos | 8 portaobjetos | |
|---|---|---|
| Agua ultrapura estéril | 50,1 | 200,4 |
| 3x hybmix | 30,0 | 120,0 |
| 1 % SDS | 0,9 | 3,6 |
| Sonda EUB 338 (100 ng/μl) | 4,5 | 18,0 |
| Sonda CMSCY301 (100 ng/μl) | 4,5 | 18,0 |
| Volumen total (μl) | 90,0 | 360,0 |
NB. Almacenar todas las soluciones que contengan oligosondas fotosensibles en la oscuridad a ‒20 °C. Proteger de la luz directa ya sea del sol o eléctrica durante su utilización.
5. Tampón fosfato 0,1 M, pH 7,0.
Na2HPO4: 8,52 g
KH2PO4: 5,44 g
Agua destilada: 1,00 l
Disolver los ingredientes, verificar el pH y esterilizar en autoclave a 121 °C durante 15 minutos.
APÉNDICE 8
Cultivo de berenjenas
Sembrar semillas de berenjena (Solanum melongena) en compost pasteurizado de semillas. Trasplantar las plántulas con los cotiledones completamente abiertos (1 a 14 días) en compost pasteurizado de tiesto.
Las berenjenas deben cultivarse en un invernadero que cumpla las siguientes condiciones ambientales:
Duración diurna: 14 horas o día natural si es de mayor duración.
Temperatura:
diurna: de 21 a 24 °C
nocturna: 15 °C
Variedades de berenjena sensibles:
'Black Beauty',
'Long Tom',
'Rima',
'Balsas'.
Proveedores: véase el sitio web: http://forum.europa.eu.int/Public/irc/sanco/Home/main
APÉNDICE 9
Procedimiento de la tinción de Gram (modificación de Hucker) (Doetsch, 1981) (1)
(1) También pueden utilizarse otras soluciones y kits de tinción disponibles en el mercado.
Solución de cristal violeta:
Disolver 2 g de cristal violeta en 20 ml de etanol al 95 %.
Disolver 0,8 g de oxalato amónico en 80 ml de agua destilada.
Mezclar las dos soluciones.
Solución de Lugol:
Yodo: 1 g
Yoduro de potasio: 2 g
Agua destilada: 300 ml
Triturar conjuntamente los sólidos en un mortero. Añadir al agua y remover hasta su disolución en un recipiente cerrado.
Solución de contraste de safranina:
Solución estándar:
SafraninaO: 2,5 g
Etanol al 95 %: 100 ml
Mezclar y conservar.
Diluir al 1:10 para conseguir una solución de trabajo.
Procedimiento de tinción:
1. Preparar los frotis, secar al aire y fijar por calor.
2. Sumergir el portaobjetos en la solución de cristal violeta durante un minuto.
3. Lavar brevemente con agua corriente.
4. Sumergir en solución de Lugol durante un minuto.
5. Lavar con agua corriente y secar.
6. Decolorar con etanol al 95 %, añadido gota a gota hasta que ya no se produzca ningún cambio de color, o sumergir agitando suavemente durante 30 segundos.
7. Lavar con agua corriente y secar.
8. Sumergir en la solución de safranina durante 10 segundos.
9. Lavar con agua corriente y secar.
Las bacterias Gram. positivas se tiñen de violeta-azul y las Gram. negativas de rosa-rojo.
BIBLIOGRAFÍA
1. Anónimo, 1987, «Scheme of the detection and diagnosis of the ring rot bacterium Corynebacterium sepedonicum in batches of potato tubers». Comisión de las Comunidades Europeas, Luxemburgo. Publ EUR 11 288 EN, 21 pp.
2. Bradbury, J. F., 1970, «Isolation and preliminary study of bacteria from plants», Rev. Pl. Path., 49, pp. 213-218.
3. Dinesen, I. G., 1984, «The extraction and diagnosis of Corynebacterium sepedonicum from diseased potato tubers», EPPO Bull. 14 (2), pp. 147-152.
4. Doetsch, R. N., 1981, «Determinative methods of light microscopy». Manual of methods for general bacteriology, American Society for Microbiology. Washington, pp. 21-23.
5. Hugh, R. F. Leifson, 1953, «The taxonomic significance of fermentative versus oxidative metabolism of carbohydrates by various gram-negative bacteria», J. Bact., 66, pp. 24-26.
6. Janse, J. D., 1991, «Infra- and intra-specific classification of Pseudomonas solanacearum strains using whole cell fattyacid análisis», Systematic and Applied Microbiology 14, pp. 335-345.
7. Janse, J. D. y J. Van Vaerenbergh, «The interpretation of the EC method for the detection of latent ring rot infections (Corynebacterium sepedonicum) in potato», EPPO Bull., 17, 1987, pp. 1-10.
8. Jansing, H. y K. Rudolph, 1998, «Physiological capabilities of Clavibacter michiganensis ssp. sepedonicus and development of a semi-selective médium», Journal of Plant Diseases and Protection, 105, pp. 590-601.
9. Klement Z.; Rudolph, K y D. C. Sands, 1990. Methods in Phytobacteriology. Akadémiai Kiadó, Budapest, 568 pp.
10. .Kovacs, N., 1956, «Identification of Pseudomonas pyocyanea by the oxidase reaction», Nature, Londres, 178, pp. 703
11. Lelliott, R. A., 1966, «The plant pathogenic coryneform bacteria», J. appl. Bact., 29, pp. 114-118.
12. Lelliott, R. A., E. Billing y A. C. Hayward, 1966, «A determinative scheme for the fluorescent plant pathogenic pseudomonads », J. appl. Bact., 29, pp. 470-489.
13. Lelliott, R. A. y P. W. Sellar, 1976, «The detection of latent ring rot [Corynebacterium sepedonicum (Spiek. et Kotth.) Skapt. et Burkh.)] in potato stocks», EPPO Bull., 6 (2), pp. 101-106.
14. Lelliott, R.A. and Stead, D.E. 1987. Methods for the diagnosis of bacterial diseases of plants. Blackwell scientific Publications Ltd., Oxford. 216 pp.
15. Li, X. y S.H. de Boer, 1995, «Selection of Polymerase Chain Reaction primers from RNA intergenic spacer región for specific detection of Clavibacter michiganensis ssp. sepedonicus», Phytopathology, 85, pp. 837-842.
16. Mills, D., Russell, B., W. y J., W. Hanus, 1997, «Specific detection of Clavibacter michiganensis ssp. sepedonicus by amplification of three unique DNA sequences isolated by subtraction hybridization», Phytopathology, 87, 8, pp. 853-861.
17. Pastrik, K. -H. y R.A. Rainey. 1999, «Identification and differentiation of Clavibacter michiganensis subspecies by polymerase chain reaction-based techniques», J. Phytopathology 147; pp. 687-693.
18. Pastrik, K.-H., 2000, «Detection of Clavibacter michiganensis ssp. sepedonicus in potato tubers by multiplex PCR with coamplification of host DNA», European Journal of Plant Pathology, 106, pp. 155-165.
19. Ramamurthi, C. S., 1959, «Comparative studies on some Gram-positive phytopathogenic bacteria and their relationship to the Corynebacteria», Mem. Cornell agrie. Exp. Sta., 366, 52 pp.
20. Schaad, W., Berthier-Schaad, Y., Sechler, A. y Knorr, D. (1999), «Detection of Clavibacter michiganensis subsp. sepedonicus in potato tubers by BIO-PCR and an automated real-time fluorescence detection system», Plant Disease 83; pp. 1095-1100.
21. Schaad, W. 2001, «Laboratory guide for identification of plant pathogenic bacteria», Schaad [Hrsg.]. 3a. ed.; St. Paul, Minnesota, 373 pp.
22. Skerman, V. B. D., 1967, A guide to the identification of the genera of bacteria. Segunda edición, Williams and Wilkins Company, Baltimore, 1967.
23. Smith, N. C.; Hennesy, J; Stead, D.E., 2001, «Repetetive sequence-derived PCR profiling using the BOX-A1 Ralstonia solanacearum primer for rapid identification of plant pathogen Clavibacter michiganensis ssp. sepedonicus», European Journal of Plant Pathology, 107 (7), pp. 739-748.
24. Sneath, P. H. A. y V. G. Collins, 1974, «A study in test reproductibility between laboratories: report of Pseudomonas working party», Antonie van Leeuwenhoek, 40, pp. 481-527.
25. Stead, D.E., 1992, «Grouping of plant pathogenic and some other Pseudomonas spp. using cellular fatty- acid profiles», International Journal of Systematic Bacteriology 42; pp. 281-295.
26. Wullings, B. A.; van Beuningen, A. R.; Janse, J. D. y A. D. L. Akkermans, 1998, «Detection of Ralstonia solanacearum, which causes brown rot of potato, by fluorescent in situ hybridization with 23s rRNA-targeted probes», Appl. Environ. Microbiol, 64, pp. 4546-4554.
1. En cada caso de foco sospechoso para el que se haya realizado una o varias pruebas de selección con resultados positivos de acuerdo con los métodos establecidos en el anexo I, y se espere recibir confirmación o refutación por aplicación de los citados métodos, se deberá proceder a la retención y adecuada conservación de:
‒ todos los tubérculos y, siempre que sea posible, las plantas de los que se hayan obtenido muestras,
‒ cualquier extracto sobrante y cualquier material adicional que se haya preparado para la prueba o pruebas de selección (por ejemplo, las placas de inmunofluorescencia),
y
‒ toda la documentación pertinente, hasta completar la aplicación de los métodos arriba mencionados.
La retención de los tubérculos permitirá que se puedan llevar a cabo pruebas de variedades cuando se estime oportuno.
2. En el caso de confirmación de la presencia del organismo, se procederá a la retención y adecuada conservación:
‒ del material especificado en el punto 1,
‒ de una muestra de las berenjenas infectadas inoculadas con extracto de tubérculo o de planta,
y
‒ del cultivo aislado del organismo,
hasta un mes después, como mínimo, en virtud del procedimiento establecido en el artículo 5, apartado 2.
1. Los elementos que deben tenerse en cuenta para la determinación de la extensión de la probable contaminación con arreglo al artículo 5, apartado 1, letra b), incluirán:
‒ los tubérculos o plantas cultivados en un lugar de producción declarado contaminado en virtud del artículo 5, apartado 1, letra a);
‒ el lugar o lugares de producción relacionados en alguna medida con la producción de los tubérculos o plantas declarados contaminados en virtud del artículo 5, apartado 1, letra a), incluido el material y las instalaciones de producción compartidos directamente o a través de un contratista común;
‒ los tubérculos o plantas producidos en el lugar o los lugares de producción contemplados en el guión anterior, o presentes en tal(es) lugar(es) de producción durante el período en el que los tubérculos o plantas declarados contaminados según el artículo 5, apartado 1, letra a), se encontraban en el lugar de producción citado en el primer guión;
‒ los locales que manipulen patatas procedentes de los lugares de producción mencionados en los guiones anteriores;
‒ cualquier maquinaria, vehículo, buque, almacén o unidades de estos y cualesquiera otros objetos, incluido el material de embalaje, que puedan haber estado en contacto con los tubérculos o plantas declarados contaminados en virtud del artículo 5, apartado 1, letra a);
‒ todos los tubérculos o plantas almacenados o que hayan estado en contacto con cualquiera de las estructuras u objetos enumerados en el guión anterior antes de la limpieza y desinfección de los mismos;
‒ como resultado de las pruebas contempladas en el artículo 6, aquellos tubérculos o plantas que tengan una relación clonal fraterna o paternal con los tubérculos o plantas declarados contaminados en virtud del artículo 5, apartado 1, letra a), y con respecto a los cuales parezca probable la contaminación a través de un vínculo clonal, aunque las pruebas de detección del organismo hayan arrojado resultados negativos; pueden llevarse a cabo pruebas de variedades para verificar la identidad de los tubérculos o plantas contaminados y clonalmente relacionados,
y
‒ el lugar o lugares de producción de los tubérculos o plantas a los que se hace referencia en el guión anterior.
2. Los elementos que deben tenerse en cuenta para la determinación de la posible propagación con arreglo al artículo 5, apartado 1, letra c), incluirán:
‒ la proximidad de otros lugares de producción en los que se cultiven patatas u otras plantas huéspedes, y
‒ la producción en común y el uso común de existencias de patatas de siembra.
3. La notificación a que se hace referencia en el artículo 5, apartado 2, se ajustará a lo siguiente:
‒ se efectuará de manera inmediata una vez que la presencia haya sido confirmada por las pruebas de laboratorio con arreglo a los métodos expuestos en el anexo I, y deberá recoger, como mínimo:
• el nombre de la variedad del lote de patatas,
• el tipo (de consumo, de siembra, etc.) y, en su caso, la categoría de patata de siembra.
‒ cuando exista el riesgo de contaminación de patatas procedentes de otra u otras Comunidades Autónomas, la Comunidad Autónoma en que se haya confirmado el brote notificará inmediatamente a las Comunidades Autónomas interesadas la información necesaria para, en su caso, declarar la contaminación, determinar la extensión de la probable contaminación y delimitar una zona de conformidad con las letras a), b), y c) del apartado 1 del artículo 5, incluyendo:
• el nombre de la variedad del lote de patatas,
• el nombre y la dirección del proveedor y del destinatario,
• la fecha de entrega del lote de patatas,
• el tamaño del lote de patatas entregado,
• una copia del pasaporte fitosanitario o al menos el número de dicho pasaporte, en su caso, o el número de registro del cultivador o del comerciante, en su caso, y una copia del aviso de entrega.
Cuando se suministre dicha información, la Comunidad Autónoma informará de ello inmediatamente al Ministerio de Agricultura, Pesca y Alimentación.
‒ cuando exista el riesgo de contaminación de patatas destinadas a otro u otros Estados Miembros, la Comunidad Autónoma en que se haya confirmado el brote notificará inmediatamente al Ministerio de Agricultura, Pesca y Alimentación y este a su vez, a través del cauce correspondiente a los Estados Miembros interesados, la información necesaria para, en su caso, declarar la contaminación y delimitar una zona de conformidad con las letras a), b), y c) del apartado 1 del artículo 5, incluyendo:
• el nombre de la variedad del lote de patatas,
• el nombre y la dirección del proveedor y del destinatario,
• la fecha de entrega del lote de patatas,
• el tamaño del lote de patatas entregado,
• una copia del pasaporte fitosanitario o al menos el número de dicho pasaporte, en su caso, o el número de registro del cultivador o del comerciante, en su caso, y una copia del aviso de entrega.
Cuando se suministre dicha información, el Ministerio de Agricultura, Pesca y Alimentación informará de ello inmediatamente a la Comisión.
‒ cuando exista el riesgo de contaminación de patatas procedentes de otro u otros Estados Miembros a raíz de la correspondiente notificación o notificaciones de los mismos, el Ministerio de Agricultura, Pesca y Alimentación comunicará la información recibida a las Comunidades Autónomas interesadas para, en su caso, declarar la contaminación, determinar la extensión de la probable contaminación y delimitar una zona de acuerdo con las letras a), b), y c) del apartado 1 del artículo 5.
‒ Una vez que todas las investigaciones hayan concluido, las Comunidades Autónomas facilitarán, para cada caso:
• la fecha en la que se confirmó la contaminación.
• una breve descripción de la investigación llevada a cabo para identificar la fuente y la posible propagación de la contaminación, incluido el alcance del muestreo efectuado,
• información sobre la fuente o fuentes de contaminación determinadas o presuntas,
• detalles relativos a la extensión de la contaminación declarada, incluido el número de lugares de producción y el número de lotes, con la indicación de la variedad y, en el caso de la patatas de siembra, la categoría,
• detalles relativos a la delimitación de la zona, incluido el número de lugares de producción no declarados contaminados pero incluidos en la zona,
• cualesquiera otros datos que requiera el Ministerio de Agricultura, Pesca y Alimentación o, en su caso, la Comisión relativos al brote o brotes confirmados.
1. Las medidas bajo control oficial contempladas en el artículo 7, letra a), serán las siguientes:
‒ la utilización como piensos tras un tratamiento térmico adecuado que garantice que no hay riesgo alguno de supervivencia del organismo,
o bien
‒ la eliminación en un vertedero autorizado oficialmente a tal fin donde no se haya podido determinar riesgo alguno de escape del patógeno al medio ambiente, por ejemplo, mediante la filtración a tierras de cultivo,
o bien
‒ la incineración,
o bien
‒ la transformación industrial mediante entrega directa e inmediata a una planta de transformación dotada de instalaciones de eliminación de residuos autorizadas oficialmente, con respecto a las cuales se haya excluido cualquier riesgo identificable de propagación del organismo, y de un sistema de limpieza y desinfección de los vehículos de transporte, al menos,
o bien
‒ otras medidas, siempre que se haya descartado el posible riesgo de propagación del organismo, las cuales deberán notificarse al Ministerio de Agricultura, Pesca y Alimentación y este a su vez, a través del cauce correspondiente, a la Comisión y a los demás Estados miembros acompañadas de su justificación.
Todo residuo restante asociado y derivado de lo anterior deberá eliminarse mediante métodos autorizados oficialmente conforme a lo dispuesto en el anexo V de la presente orden.
2. La utilización o la eliminación apropiadas de los tubérculos o plantas declaradas probablemente contaminadas de conformidad con el artículo 5, apartado 1, letra b), a las que se hace referencia en el artículo 7, letra b), deberán efectuarse bajo el control de los organismos oficiales responsables interesados, así como con la oportuna comunicación entre estos organismos para garantizar en todo momento dicho control, y con la aprobación del organismo oficial responsable de la Comunidad Autónoma donde vayan a envasarse o transformarse las patatas, en lo que se refiere a los vertederos citados en los guiones primero y segundo, y consistirán en lo siguiente:
‒ la utilización como patatas de consumo destinadas al consumo, envasadas para su distribución y venta sin cambio de envase, en un lugar dotado de instalaciones de eliminación de residuos adecuadas; las patatas destinadas a la siembra sólo pueden manipularse en el mismo lugar si esto se realiza separadamente o tras la limpieza y desinfección,
o
‒ su uso como patatas de consumo para la transformación industrial y destinadas a la entrega directa e inmediata a una planta de transformación dotada de instalaciones adecuadas de eliminación de residuos y de un sistema de limpieza y desinfección de, al menos, los vehículos de transporte,
o
‒ algún otro tipo de uso o eliminación, siempre que se excluya todo riesgo identificable de propagación del organismo, y previa aprobación de los citados organismos oficiales responsables.
3. Los métodos apropiados de limpieza y desinfección de todos los objetos contemplados en el artículo 7, letra c), serán aquellos para los que se haya excluido cualquier riesgo identificable de propagación del organismo, y se aplicarán bajo el control de los organismos oficiales responsables de las Comunidades Autónomas.
4. La serie de medidas que deben adoptar las Comunidades Autónomas en la zona delimitada de conformidad con el artículo 5, apartado 1, letra c), a la que se hace referencia en el artículo 7, letra d), incluirá las medidas siguientes:
4.1 En los lugares de producción que se declaren contaminados en virtud del artículo 5, apartado 1, letra a):
a) en los campos que se declaren contaminados según el artículo 5, apartado 1, letra a), o bien,
i) ‒ al menos durante los tres años de cultivo siguientes al de declaración de la contaminación:
• se adoptarán medidas para eliminar las plantas de patata espontáneas y otras plantas que puedan contener naturalmente el organismo,
y
• no se plantarán tubérculos, plantas ni semillas de patata propiamente dichas, ni ninguna otra planta que pueda contener naturalmente el organismo, ni ningún cultivo que presente un riesgo cierto de propagación del organismo,
‒ en la primera temporada de cultivo de patatas siguiente al período indicado en el guión anterior y, siempre que en las inspecciones oficiales se haya comprobado que, durante al menos los dos años de vegetación inmediatamente anteriores a la plantación, el campo estuvo libre de plantas de patata espontáneas y otras plantas que puedan contener naturalmente el organismo, sólo se permitirá la producción de patatas de consumo y los tubérculos recolectados se someterán a prueba conforme al procedimiento detallado en el anexo I,
‒ en la temporada de cultivo de patatas siguiente a la mencionada en el guión anterior y tras un ciclo de rotación adecuado, cuya duración mínima será de dos años cuando vayan a cultivarse patatas de siembra, se podrán plantar patatas de siembra o de consumo y se realizará una encuesta oficial tal como se dispone el artículo 2, apartado 1, o bien,
ii) ‒ durante los cuatro años de cultivo siguientes al de declaración de la contaminación:
• se adoptarán medidas para eliminar las plantas de patata espontáneas y otras plantas que puedan contener naturalmente el organismo,
y
• se dejará y mantendrá el campo, o bien en barbecho completo, o bien en pasto permanente con siega o pastoreo intenso o frecuente,
‒ en la primera temporada de cultivo de patatas siguiente al período indicado en el guión anterior y, siempre que en las inspecciones oficiales se haya comprobado que, durante al menos los dos años de vegetación inmediatamente anteriores a la plantación, el campo estuvo libre de plantas de patata espontáneas y otras plantas que puedan contener naturalmente el organismo, se permitirá la producción de patatas de siembra o consumo y los tubérculos recolectados se someterán a prueba conforme al procedimiento detallado en el anexo I;
b) en el resto de los campos del lugar de producción contaminado, y a condición de que los organismos oficiales competentes tengan la certeza de que se ha eliminado el riesgo de plantas de patata espontáneas y de cualquier otra planta que pueda contener naturalmente el organismo:
‒ durante el año de cultivo siguiente al de declaración de contaminación o bien no se plantarán tubérculos, plantas ni semillas propiamente dichas de patata, ni cualquier otra planta que pueda contener naturalmente el organismo, o se podrán plantar patatas de siembra certificadas, pero sólo para la producción de patatas de consumo, y
‒ durante el segundo año de cultivo siguiente al de declaración de contaminación sólo se podrán plantar patatas de siembra certificadas o patatas de siembra que hayan sido sometidas a pruebas oficiales para determinar la ausencia de necrosis bacteriana y cultivadas bajo control oficial en lugares de producción distintos a los mencionados en el punto 4.1, ya sea para la producción de siembra o de consumo,
‒ durante el tercer año de cultivo siguiente al de declaración de contaminación, al menos, sólo se podrán plantar patatas de siembra certificadas o patatas de siembra cultivadas bajo control oficial a partir de patatas de siembra certificadas, ya sea para la producción de siembra o de consumo,
‒ en cada uno de los años de cultivo mencionados en los guiones anteriores, se tomarán medidas para eliminar las plantas de patata espontáneas y cualquier otra planta que pueda contener naturalmente el organismo en caso de advertirse su presencia, y se realizarán pruebas oficiales de las patatas recolectadas conforme al procedimiento detallado en el anexo I en todos los campos de patatas;
c) inmediatamente después de la declaración de contaminación en virtud del artículo 5, apartado 1, letra a), y tras el primer año de cultivo siguiente, todas las máquinas e instalaciones de almacenamiento que se encuentren en el lugar de producción e intervengan en la producción de patatas serán limpiadas y desinfectadas como resulte adecuado con los métodos apropiados, según lo dispuesto en el punto 3;
d) en el caso de las unidades de producción de cultivos protegidos en las que sea posible una sustitución total de los medios de cultivo,
‒ no se plantarán tubérculos, plantas ni semillas propiamente dichas hasta que la unidad de producción no haya sido sometida a medidas bajo control oficial destinadas a eliminar el organismo y a retirar todo el material vegetal que pueda ser huésped del mismo, incluida, como mínimo, la sustitución total del medio de cultivo y la limpieza y desinfección de la unidad de producción y de todos los equipos, y los organismos oficiales responsables no hayan concedido la subsiguiente autorización para la producción de patata,
y
‒ la producción de patata procederá de patatas de siembra certificadas o de minitubérculos o micro-plantas derivadas de fuentes probadas.
4.2 Dentro de la zona delimitada, sin perjuicio de lo dispuesto en el punto 4.1, las Comunidades Autónomas aplicarán las siguientes medidas:
a) inmediatamente después de la declaración de contaminación, velarán por que toda la maquinaria e instalaciones de almacenamiento de la explotación que hayan intervenido en la producción de patatas se limpien y desinfecten como resulte adecuado y con los métodos apropiados, según lo dispuesto en el punto 3,
b) inmediatamente y durante al menos tres temporadas de cultivo después de la declaración de contaminación:
‒ controlarán a través de sus organismos oficiales responsables las explotaciones que cultiven, almacenen o manipulen tubérculos de patata, además de las explotaciones que contraten máquinas para este cultivo,
‒ requerirán que, para todos los cultivos de patata dentro de la zona delimitada, se planten exclusivamente semillas certificadas o semillas cultivadas bajo control oficial, y se efectúe un análisis después de recolectar los cultivos de patatas de siembra en lugares de producción declarados probablemente contaminados según el artículo 5, apartado 1, letra b),
‒ exigirán que se manipulen por separado las existencias de patatas de siembra y de patatas de consumo recolectadas en todas las explotaciones de la zona, o que se establezca un sistema de limpieza y desinfección entre el manejo de las existencias de patatas de siembra y el de las de consumo.
‒ llevarán a cabo una encuesta oficial según lo dispuesto en el artículo 2, apartado 1;
c) establecerán un programa, según corresponda, para la sustitución de todas las existencias de patatas de siembra por un período de tiempo conveniente.
Los métodos oficialmente autorizados para la eliminación de residuos mencionados en el punto 1 del anexo IV se ajustarán a las disposiciones siguientes, de tal forma que se excluya todo riesgo identificable de propagación del organismo:
i) Los residuos de patata (incluidas las patatas descartadas y las mondaduras) y cualquier otro residuo sólido asociado a las patatas (incluida la tierra, las piedras y otros restos) se eliminarán de una de las siguientes maneras:
‒ en un vertedero autorizado oficialmente a tal fin donde no se haya podido determinar riesgo alguno de escape del organismo al medio ambiente, por ejemplo, mediante la filtración a tierras de cultivo; los residuos se trasladarán directamente al vertedero en unas condiciones de confinamiento que impidan todo riesgo de pérdida de los mismos,
‒ se incinerarán,
o
‒ se someterán a otras medidas, siempre que se descarte el posible riesgo de propagación del organismo; dichas medidas deben notificarse inmediatamente al Ministerio de Agricultura, Pesca y Alimentación y este a su vez, a través del cauce correspondiente, a la Comisión y a los demás Estados miembros.
ii) Los residuos líquidos que contengan elementos sólidos en suspensión se filtrarán o se someterán a procesos de sedimentación para la separación de dichos elementos; estos se eliminarán de la forma dispuesta en el inciso i).
A continuación, los residuos líquidos se someterán a alguna de las medidas siguientes:
‒ se calentarán a un mínimo de 60 °C en todo su volumen durante 30 minutos por lo menos antes de la eliminación,
o
‒ se eliminarán por otro sistema que, aprobado oficialmente y aplicado bajo control oficial, descarte todo riesgo identificable de contacto de los residuos con tierras de cultivo. Las características de tal sistema serán notificadas al Ministerio de Agricultura, Pesca y Alimentación y este a su vez, a través del cauce correspondiente, a los demás Estados miembros y a la Comisión.
Las opciones descritas en el presente anexo se aplican asimismo a los residuos asociados a la manipulación, la eliminación y el tratamiento de lotes contaminados.»
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




