Agencia Estatal Boletín Oficial del Estado






El Real Decreto 1662/2000, de 29 de septiembre, sobre productos sanitarios para diagnóstico in vitro, que incorporó al ordenamiento jurídico nacional la Directiva 98/79/CE, establece en su artículo 8.4 que se presumirá la conformidad con los requisitos esenciales, previstos en su artículo 5, de los productos diseñados y fabricados con arreglo a las especificaciones técnicas comunes elaboradas para los productos de la lista A del anexo II del mismo.
La Comisión Europea adoptó, el pasado 7 de mayo de 2002, la Decisión 2002/364/CE sobre especificaciones técnicas comunes para productos sanitarios para diagnóstico in vitro, aplicables a los productos sanitarios para diagnóstico in vitro de la lista A del anexo II de la Directiva 98/79/CE. Teniendo en cuenta que, según se establece en el artículo 8.6 del mencionado Real Decreto 1662/2000, como norma general y salvo razones justificadas, los fabricantes deberán respetar las especificaciones técnicas comunes, mediante la presente Resolución se hacen públicas las especificaciones técnicas comunes adoptadas por la Comisión Europea en su Decisión 2002/364/CE, de 7 de mayo de 2002.
En su virtud, resuelvo:
Dar publicidad a las especificaciones técnicas comunes sobre productos sanitarios para diagnóstico in vitro, adoptadas por la Comisión Europea en su Decisión 2002/364/CE, cuyo texto integro se incluye como anexo de la presente Resolución
Dichas especificaciones se adoptan como especificaciones técnicas comunes para productos sanitarios para diagnóstico in vitro de la lista A del anexo II del Real Decreto 1662/2000, de 29 de septiembre.
Los fabricantes deberán, como norma general, respetar las citadas especificaciones técnicas comunes. Si por razones debidamente justificadas, los fabricantes no cumplieran estas especificaciones deberán adoptar soluciones de un nivel al menos equivalente a las mismas.
Madrid, 14 de abril de 2003.–El Subsecretario, Pablo Vázquez Vega.
1. Ámbito de aplicación
Las presentes especificaciones técnicas comunes son aplicables a los productos recogidos en la lista A del anexo II:
Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la determinación de los grupos sanguíneos siguientes: sistema ABO, Rhesus (C, c, D, E, e) y anti-Kell.
Reactivos y productos reactivos, incluidos los materiales asociados de calibrado y control, para la detección, confirmación y cuantificación en muestras humanas de marcadores de infección por VIH (VIH 1 y VIH 2), HTLV I y II, y de hepatitis B, C y D.
2. Definiciones
Sensibilidad (diagnóstica). La probabilidad de que el producto dé un resultado positivo en presencia de un marcador diana.
Verdadero positivo. Una muestra conocida como positiva para el marcador diana y correctamente clasificada por el producto.
Falso negativo. Una muestra conocida como positiva para el marcador diana e incorrectamente clasificada por el producto.
Especificidad (diagnóstica). La probabilidad de que un producto dé un resultado negativo en ausencia de un marcador diana.
Falso positivo. Una muestra conocida como negativa para el marcador diana e incorrectamente clasificada por el producto.
Verdadero negativo. Una muestra conocida como negativa para el marcador diana y correctamente clasificada por el producto.
Sensibilidad analítica. En el contexto de las ETC puede expresarse como el límite de detección: la cantidad más pequeña del marcador diana que puede ser detectada con precisión.
Especificidad analítica. La capacidad del método para determinar solamente el marcador diana.
Técnicas de amplificación de ácidos nucleicos (NAT). En el contexto de este documento el término «NAT» es utilizado para las pruebas de detección y/o cuantificación de ácidos nucleicos ya sea por amplificación de una secuencia objetivo, por amplificación de una señal o por hibridación. Prueba rápida. En este contexto el término «prueba rápida» se entiende como aquellas pruebas que sólo pueden ser utilizadas individualmente o en una serie corta y que han sido diseñadas para proporcionar un resultado inmediato a la cabecera del paciente.
Consistencia. La consistencia de un procedimiento de análisis es una medida de su capacidad para no ser afectada por las variaciones pequeñas pero deliberadas de los parámetros del método, y proporciona una indicación de su fiabilidad durante el uso normal.
Tasa de fallo del sistema completo. La tasa de fallo del sistema completo es la frecuencia de fallos cuando el proceso completo se realiza según las indicaciones del fabricante.
3. Especificaciones técnicas comunes (ETC) para productos definidos en la lista a del anexo II de la Directiva 98/79/CE
3.1 ETC para la evaluación de funcionamiento de reactivos y productos reactivos para la detección, confirmación y cuantificación en muestras humanas de marcadores de infección por VIH (VIH 1 y VIH 2), HTLV I y II, y hepatitis B, C y D.
Principios generales.
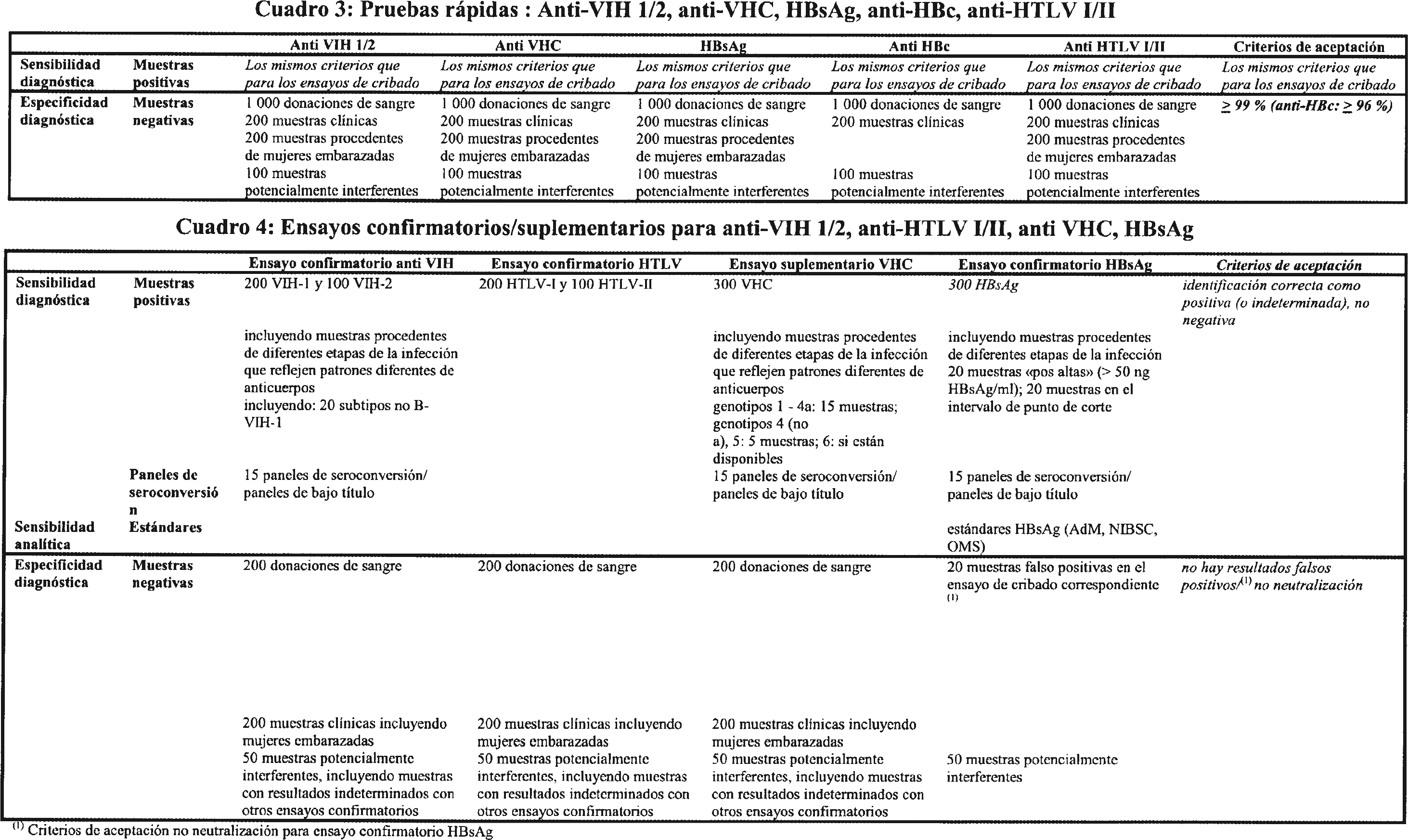
3.1.1 Los productos para la detección de infecciones virales deberán cumplir los mismos requisitos de sensibilidad y especificidad tanto si son comercializados para el cribado de muestras como si lo son para diagnóstico (véase el cuadro 1).
3.1.2 Los productos que los fabricantes destinen para utilizar en fluidos corporales que no sean suero o plasma, como por ejemplo, orina, saliva, etc., cumplirán los mismos requisitos de sensibilidad y especificidad de las ETC que los ensayos para suero y plasma. En la evaluación de funcionamiento se analizarán muestras de los mismos individuos tanto en el ensayo que deberá ser aprobado como en un ensayo análogo para suero o plasma.
3.1.3 Los productos que los fabricantes destinen para autodiagnóstico, es decir, para uso doméstico, cumplirán los mismos requisitos de sensibilidad y especificidad de las ETC que sus productos análogos para uso profesional. Las fases relevantes de la evaluación de funcionamiento se realizarán (o repetirán) por usuarios legos con el fin de validar el funcionamiento del producto y las instrucciones de uso.
3.1.4 Todas las evaluaciones de funcionamiento se realizarán en comparación directa con un producto establecido cuyo funcionamiento sea aceptable. El producto de comparación utilizado deberá tener el marcado CE, si está comercializado en el momento de realizar la evaluación de funcionamiento.
3.1.5 Si se identifican resultados discrepantes de un ensayo durante una evaluación, deberán resolverse hasta donde sea posible, por ejemplo:
evaluando la muestra discrepante por sistemas de ensayo adicionales utilizando métodos o marcadores alternativos
revisando el estado clínico y el diagnóstico del paciente y,
analizando muestras de seguimiento.
3.1.6 Las evaluaciones de funcionamiento se realizarán sobre una población equivalente a la población europea.
3.1.7 Las muestras positivas utilizadas en la evaluación de funcionamiento se seleccionarán para reflejar las diferentes etapas de la enfermedad o enfermedades de que se trate, diferentes patrones de anticuerpos, diferentes genotipos, diferentes subtipos, etc.
3.1.8 En el caso de productos para el cribado de sangre (a excepción de los ensayos para la determinación del HBsAg), todas las muestras verdaderas positivas serán identificadas como positivas por el producto que deba recibir el marcado CE (cuadro 1). En el caso de los ensayos para HBsAg, el nuevo producto tendrá unos resultados globales al menos equivalentes a los del producto establecido (véase el principio 3.1.4). La sensibilidad diagnóstica del ensayo durante la fase de infección temprana (seroconversión) debe estar al nivel del estado actual de la técnica. El reanálisis de los mismos paneles o paneles adicionales de seroconversión, ya sea realizado por el organismo notificado o por el fabricante, confirmará los resultados iniciales de la evaluación de funcionamiento (véase el cuadro 1).
3.1.9 Las muestras negativas utilizadas en la evaluación de funcionamiento reflejarán la población diana del ensayo, por ejemplo, donantes de sangre, pacientes hospitalizados, mujeres embarazadas, etc.
3.1.10 Para la evaluación de funcionamiento de ensayos de cribado (cuadro 1), las poblaciones de donantes de sangre investigadas procederán de al menos dos centros de donación y deberán provenir de donaciones de sangre consecutivas no seleccionadas para excluir muestras de individuos que donan por primera vez.
3.1.11 Los productos tendrán una especificidad de al menos el 99,5 % en donantes de sangre, si no se indica lo contrario en los cuadros adjuntos. La especificidad se calculará mediante la frecuencia de resultados repetidamente reactivos (esto es, falsos positivos) en donantes de sangre negativos para el marcador diana.
3.1.12 Durante la evaluación de funcionamiento, los productos se evaluarán para establecer el efecto de sustancias potencialmente interferentes. Estas sustancias potencialmente interferentes dependerán en cierto modo de la composición del reactivo y la configuración del ensayo. Las sustancias potencialmente interferentes se identificarán como parte del análisis de riesgos exigido en los requisitos esenciales para cada nuevo producto pero podrán incluir, por ejemplo:
Muestras que representan infecciones «relacionadas».
Muestras procedentes de mujeres embarazadas multíparas, esto es, que han tenido más de un embarazo, o pacientes positivos para el factor reumatoide.
En el caso de antígenos recombinantes, muestras con anticuerpos humanos a componentes del sistema de expresión utilizado para los antígenos recombinantes, por ejemplo anti E. coli o anti levadura.
3.1.13 Para productos destinados por el fabricante a su uso en suero y plasma, la evaluación de funcionamiento debe demostrar la equivalencia entre suero y plasma. Esto se demostrará para 50 donaciones, como mínimo.
3.1.14 Para los productos destinados a su uso en plasma, la evaluación de funcionamiento verificará el funcionamiento del producto utilizando todos los anticoagulantes que el fabricante indique aptos para emplearse con el producto. Esto se demostrará para 50 donaciones, como mínimo.
3.1.15 Como parte del análisis de riesgos exigido, se determinará la tasa de fallo completo del sistema que genera resultados falsos negativos mediante ensayos repetidos en muestras positivas débiles.
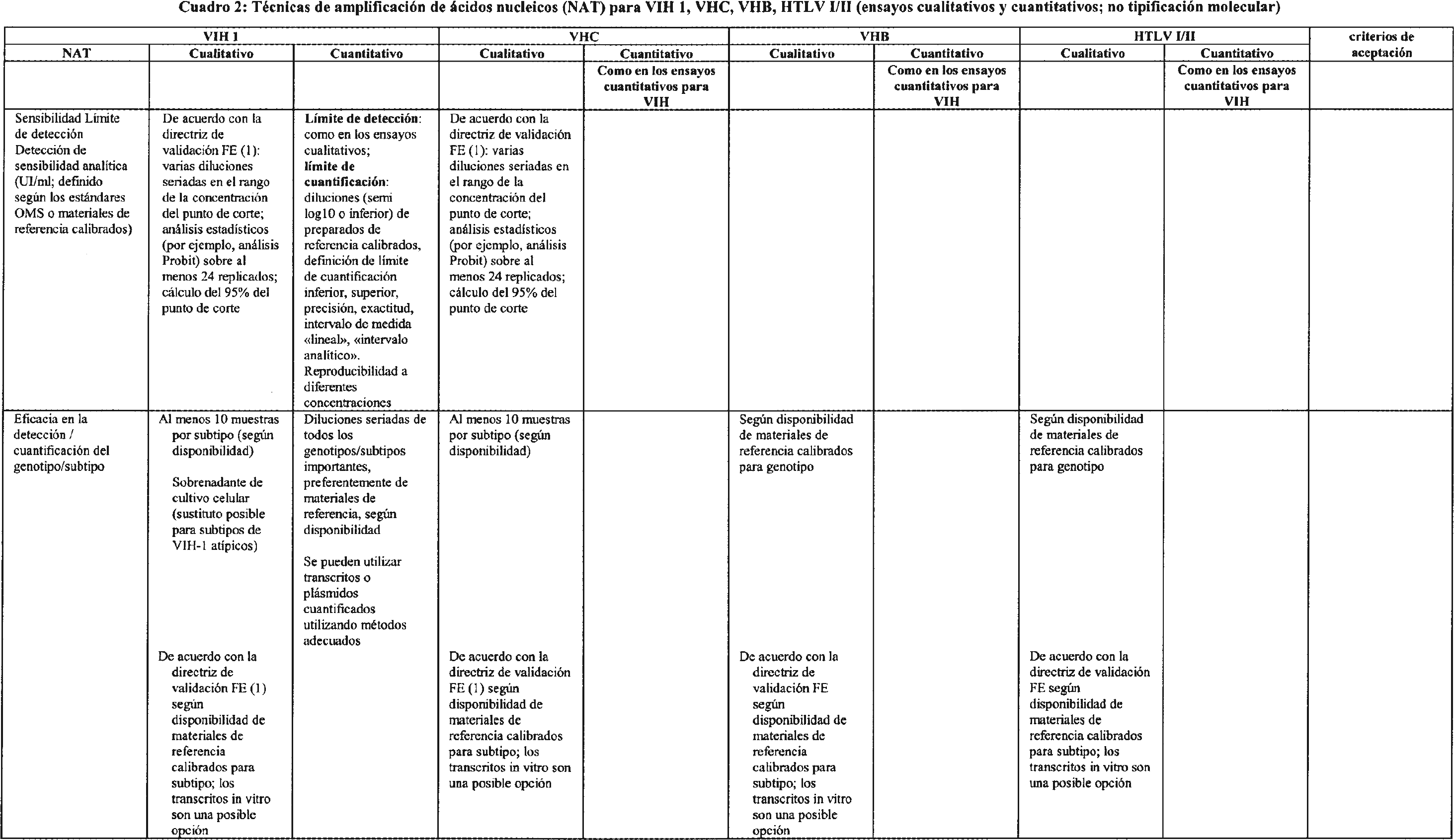
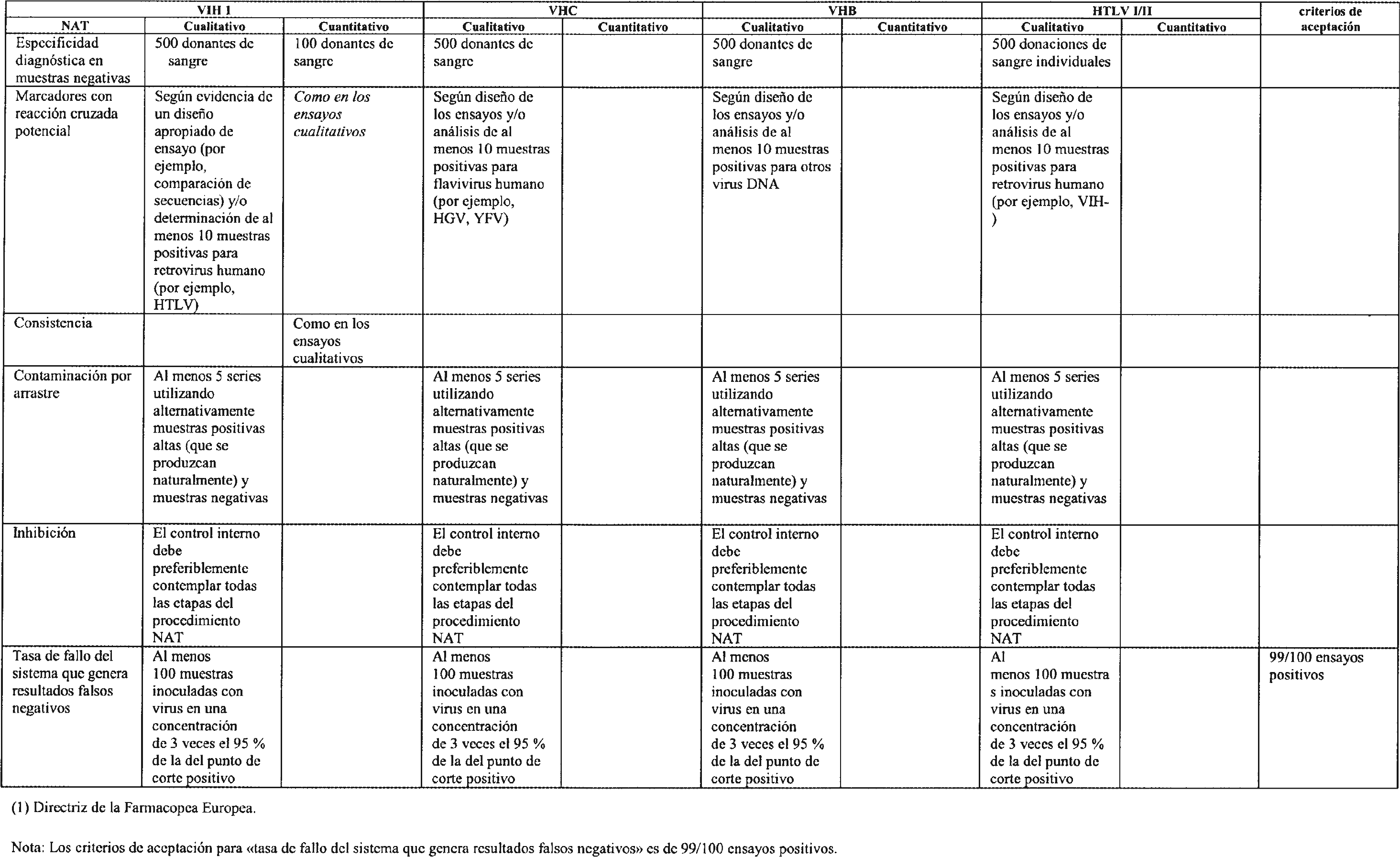
3.2 Requisitos adicionales para técnicas de amplificación de ácidos nucleicos (NAT). Los criterios de evaluación de funcionamiento para los ensayos NAT pueden verse en el cuadro 2.
3.2.1 En el caso de los ensayos de amplificación de una secuencia diana, la inclusión de un control de funcionalidad para cada muestra ensayada (control interno) reflejará el estado actual de la técnica. Hasta donde sea posible, este control se utilizará durante todo el proceso, esto es, extracción, amplificación/hibridación y detección.
3.2.2 La sensibilidad analítica o límite de detección de un ensayo NAT se expresará como el 95 % del punto de corte positivo. Esta es la concentración del analito para la que el 95 % de las series de ensayo dan resultados positivos tras diluciones seriadas de un material de referencia internacional, por ejemplo un estándar de la OMS, o materiales de referencia calibrados.
3.2.3 La detección del genotipo se demostrará mediante la adecuada validación del diseño de la sonda y el cebador, y también se validará ensayando muestras con genotipo caracterizado.
3.2.4 Los resultados de los ensayos NAT cuantitativos serán trazables a estándares internacionales o materiales de referencia calibrados, si existen, y se expresarán en las unidades internacionales utilizadas en el ámbito específico de aplicación.
3.2.5 Los ensayos NAT podrán utilizarse para detectar virus en muestras negativas para anticuerpos, esto es, muestras previas a la seroconversión. Los virus incluidos en los inmunocomplejos pueden comportarse de forma diferente a los virus libres, por ejemplo durante la centrifugación. Por tanto, es importante que en las evaluaciones de consistencia se incluyan muestras negativas para anticuerpos (muestras previas a la seroconversión).
3.2.6 Para el estudio de la contaminación por arrastre, en los estudios de consistencia se analizarán al menos cinco series alternando muestras positivas altas y muestras negativas. Las muestras positivas altas serán muestras con títulos altos que se generen de forma natural.
3.2.7 La tasa de fallo completo del sistema que genera resultados falsos negativos se determinará analizando muestras positivas débiles. Las muestras positivas débiles deberán contener una concentración de virus equivalente a 3 veces el 95 % del punto de corte positivo de concentración del virus.
3.3 ETC para la aprobación por el fabricante de reactivos y productos reactivos para la detección, confirmación y cuantificación en muestras humanas de marcadores de infección por VIH (VIH 1 y VIH 2), HTLV I y II, y hepatitis B, C y D (ensayos inmunológicos solamente).
3.3.1 El criterio de aprobación por el fabricante garantizará que cada lote identifica de manera constante los antígenos, epítopos y anticuerpos correspondientes.
3.3.2 Se incluirán al menos 100 muestras negativas para el analito en cuestión en los ensayos de aprobación de lotes de los fabricantes.
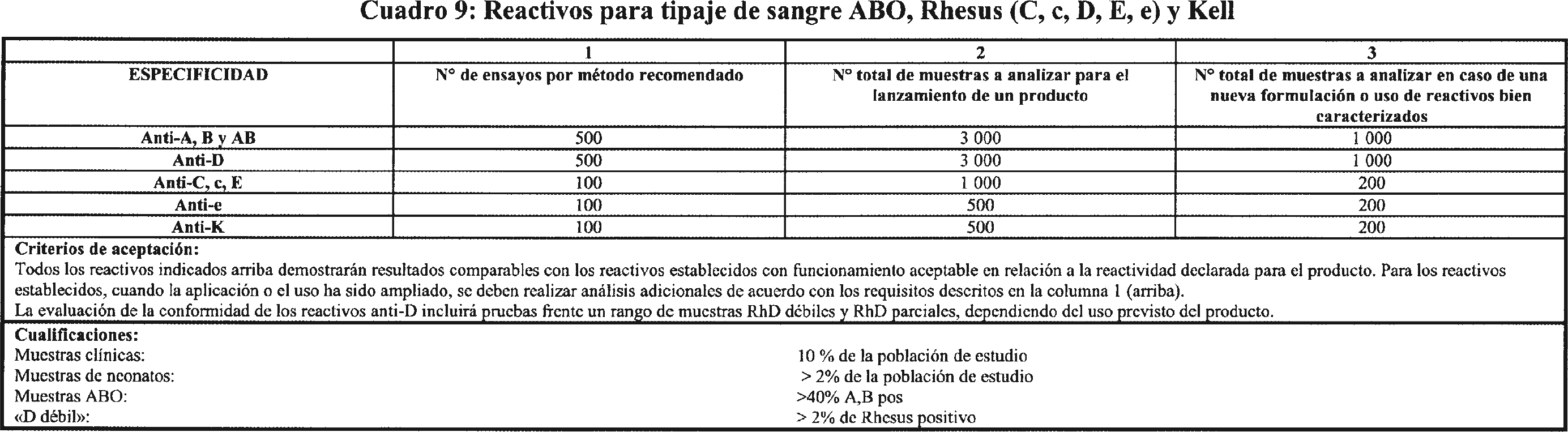
3.4 ETC para la evaluación del funcionamiento de reactivos y productos reactivos para la determinación de los grupos sanguíneos: sistema ABO (A,B), Rhesus (C, c, D, E, e) y Kell (K). Los criterios para la evaluación de funcionamiento de reactivos y productos reactivos para la determinación de los grupos sanguíneos: sistema ABO (A,B), Rhesus (C, c, D, E, e) y Kell (K) se indican en el cuadro 9.
3.4.1 Todas las evaluaciones de funcionamiento se realizarán en comparación directa con un producto establecido cuyo funcionamiento sea aceptable. El producto de comparación utilizado debe tener el marcado CE, si está comercializado en el momento de realizar la evaluación de conformidad.
3.4.2 Si se identifican resultados discrepantes de un ensayo durante una evaluación, deberán resolverse hasta donde sea posible, por ejemplo:
Evaluando la muestra discrepante por sistemas de ensayo adicionales.
Utilizando un método alternativo.
3.4.3 Las evaluaciones de funcionamiento se realizarán sobre una población equivalente a la población europea.
3.4.4 Las muestras positivas utilizadas para la evaluación de funcionamiento se seleccionarán para reflejar la expresión de antígenos variantes y débiles.
3.4.5 Durante la evaluación de funcionamiento, los productos se evaluarán para establecer el efecto de sustancias potencialmente interferentes. Estas sustancias potencialmente interferentes dependerán en cierto modo de la composición del reactivo y la configuración del ensayo. Las sustancias potencialmente interferentes se identificarán como parte del análisis de riesgos exigido en los requisitos esenciales para cada nuevo producto.
3.4.6 Para los productos destinados a su uso en plasma, la evaluación de funcionamiento verificará el funcionamiento del producto utilizando todos los anticoagulantes que el fabricante indique aptos para emplearse con el producto. Esto se demostrará para 50 donaciones, como mínimo.
3.5 ETC para la aprobación por el fabricante de reactivos y productos reactivos para la determinación de antígenos de los grupos sanguíneos: sistema ABO (A, B), Rhesus (C, c, D, E, e) y Kell (K).
3.5.1 El criterio de aprobación por el fabricante garantizará que cada lote identifica de manera constante los antígenos, epítopos y anticuerpos correspondientes.
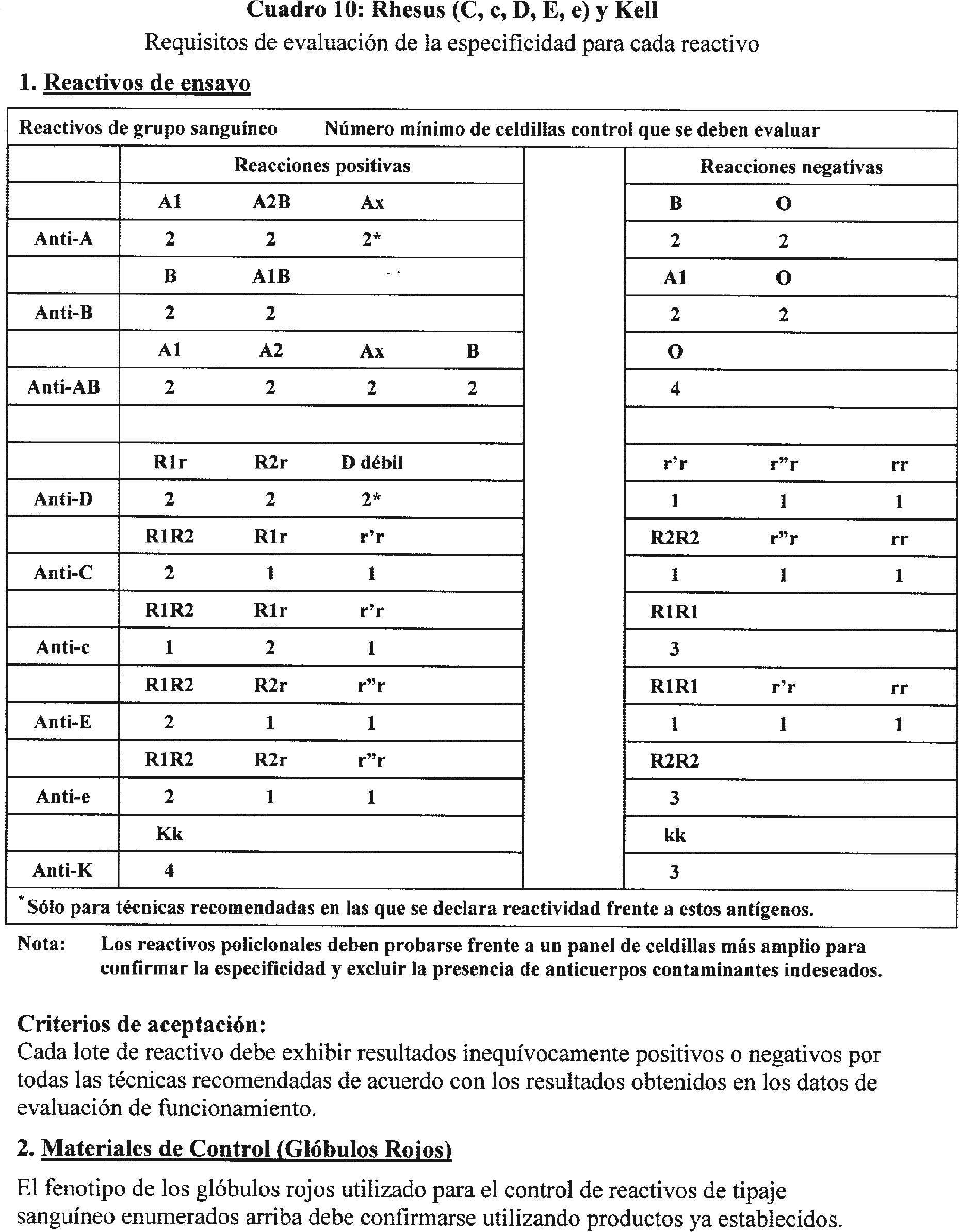
3.5.2 Los requisitos de aprobación por lotes por el fabricante se describen en el cuadro 10.



Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




