Agencia Estatal Boletín Oficial del Estado






Excelentísimos señores:
De conformidad con lo establecido en las Ordenes de 31 de enero de 1977 («Boletín Oficial del Estado» de 14 de julio), 31 de julio de 1979 («Boletín Oficial del Estado» de 20 y 30 de agosto), 17 de septiembre de 1981 («Boletín Oficial del Estado» de 14 de octubre) y 1 de diciembre de 1981 («Boletín Oficial del Estado» de 20 de enero de 1982), por las que se declaraban como oficiales los diversos métodos de análisis, se ha continuado ensayando y poniendo a punto los nuevos métodos, no sólo de las materias comprendidas en las citadas Ordenes, sino de otras nuevas donde existe necesidad urgente de disponer de métodos de análisis para el control de determinados parámetros analíticos.
En su virtud, a propuesta de los Ministerios de Economía y Hacienda, de Industria y Energía, de Agricultura, Pesca y Alimentación y de Sanidad y Consumo, previo informe preceptivo de la Comisión Interministerial para la Ordenación Alimentaria y oídos los representantes de las Organizaciones profesionales afectadas, esta Presidencia del Gobierno dispone:
Se aprueba como oficiales los métodos de análisis del aceite y grasas que se citan en el anexo I.
Cuando no existan métodos oficiales para determinados análisis, y hasta tanto los mismos no sean propuestos por el órgano competente y previamente informados por la Comisión Interministerial para la Ordenación Alimentaria, podrán ser utilizados los adoptados por los Organismos nacionales e internacionales de reconocida solvencia.
A la entrada en vigor de la presente Orden queda derogado el método 24a de la Orden de 31 de enero de 1977 («Boletín Oficial del Estado» de 15 de julio), y cuantas otras disposiciones de igual o inferior rango se opongan a lo establecido en la misma.
La presente disposición entrará en vigor a los treinta días de su publicación en el «Boletín Oficial del Estado».
Madrid, 9 de octubre de 1985.
MOSCOSO DEL PRADO Y MUÑOZ.
Excmos. Sres. Ministros de Sanidad y Consumo, de Economía y Hacienda, de Industria y Energía y de Agricultura, Pesca y Alimentación.
‒ 24(a). Esteroles.
‒ 54. Actividad de adsorbentes para cromatografía en columna.
‒ 55. Ceras en aceite de girasol.
‒ 56. Tocoferoles.
‒ 57. Aceites semisecantes en aceites de oliva y orujo.
‒ 58. Determinación del eritrodiol.
24(a). Esteroles
24(a).1 Principio.
1) Saponificación de la grasa y extracción de la materia insaponificable.
2) Aislamiento de la fracción de esteroles por orometografía en capa fina.
3) Separación de los componentes de la fracción de esteroles por cromatografía gaseosa.
24(a).2 Material y aparatos.
24(a).2.1 Material descrito en la norma 22(b) ‒insaponificable‒ método del éster etílico.
24(a).2.2 Equipo de preparación de las placas para cromatografía en capa fina.
24(a).2.3 Placas de vidrio de 20 x 20 x 0,4 cm.
24(a).2.4 Cubeta de vidrio, con su tapa, para el desarrollo de las placas.
24(a).2.5 Pulverización para la aplicación del reactivo de revelado a las placas.
24(a).2.6 Cromatógrafo de gas, con un sistema de detección sensible, preferiblemente de ionización de llama de hidrógeno, provisto de inyector de vidrio.
24(a).2.7 Columna de vidrio de uno dos m de longitud (diámetro exterior siete mm; diámetro interior dos mm), con relleno al 3 por 100 de una metilsilicona o una metil-fenil-silicona (SE-30, OV-17 son adecuadas), sobre Gas-Chrom Q, Chromosrob W-HP o Supelcoport, con una granulometría de 80-100 mallas.
24(a).2.8 Matraces en forma de corazón de 10 mm, provistos de tapones esmerilados.
24(a).2.9 Microjeringa de 5 o 10 µl, graduada en 0,1 o 0,2 µl.
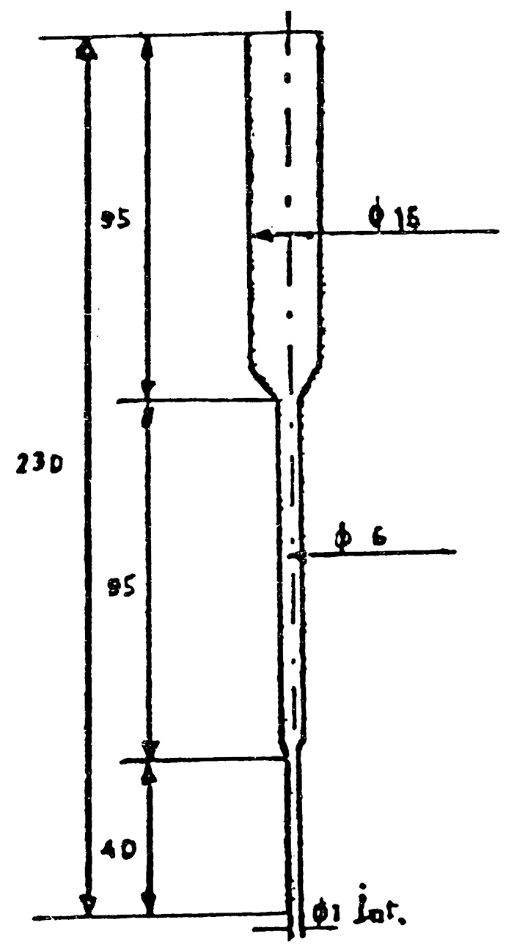
24(a).2.10 Columna cromatográfica destinada a la elución de las fracciones separadas por cromatografía en capa fina, tal como se indica en la figura.
24(a).3 Reactivos.
24(a).3.1 Reactivos descritos en la norma 22(b) ‒insaponificable‒ método del éster etílico.
24(a).3.2 Gel de sílice G.
24(a).3.3 Rodamina 6G al 0,001 por 100 en etanol del 90 por 100. Se puede emplear cualquier otro colorante que no reaccione con los esteroles, como por ejemplo diclorofluoresceína.
24(a).3.4 Cloroformo.
24(a).3.5 Colesterol purísimo, disolución al 1 por 100 en cloroformo.
24(a).3.6 Hexanol.
24(a).3.7 Líquido de desarrollo para la cromatografía en capa fina. Preparar una mezcla hexano-éter etílico 60:40 (v/v), a la que se agrega 1 por 100 de ácido fórmico.
24(a).4 Procedimiento.
24(a).4.1 Saponificación de la grasa y extracción del insaponificable.
Extraer el insaponificable de cinco g de muestra, según el método 22(b) «método de éter etílico» siguiendo la marcha descrita y eliminar el disolvente volátil en evaporador rotatorio, cuidando de no calentar a temperatura superior a 50 ºC.
Disolver el insaponificable en dos ml de cloroformo.
24(a).4.2 Aislamiento de fracción de esteroles por cromatografía en capa fina.
24(a).4.2.1 Preparación de las cromatoplacas. Antes de proceder a la extensión de la pasta de gel de sílice, limpiar las placas con hexano, etanol y acetona sucesivamente.
En un frasco de unos 250 ml de capacidad con cierre hermético y tapón de rosca, añadir gel de sílice y aproximadamente el doble de su peso de agua. Ajustar el tapón y agitar enérgicamente durante un minuto, 28 g del gel de sílice son suficientes para la preparación de cuatro placas de 20 x 20 cm. Se pasa la masa al aplicador y se extiende sobre las placas con un espesor desde la mezcla de gel con agua hasta su extensión que se efectuará de una forma continua para lograr la mayor homogeneidad de la capa de sílice.
Dejar secar las placas al aire unos treinta minutos e introducirlas en estufa a 105 ºC durante una hora. Guardar en desecador.
24(a).4.2.2 Preparación de la cubeta. Introducir en la cubeta el líquido de desarrollo, mezcla hexano-éter 60-40 hasta una altura de un cm, tapar y dejar en reposo durante tres horas, antes de su utilización, con el fin de lograr el equilibrio líquido-vapor en el interior de la cubeta.
24(a).4.2.3 Cromatografía en capa fina. Depositar 1 ml del insaponificable, en una banda, a distancia de 2 cm del borde inferior y a 2,5 cm de los bordes laterales a la misma altura y a 1 cm de los bordes depositar 30µl de patrón de colesterol al 1 por 100.
Introducir la placa en la cubeta, tapar y esperar hasta que el desarrollo del frente líquido esté situado a una distancia aproximada de 1 cm del límite superior de la placa.
Secar la placa y dejarla secar a temperatura ambiente. Pulverizar las manchas patrón con el líquido de revelado y localizar la posición de la banda de esteroles con lámpara de UV a 366 nm.
Rascar la sílice correspondiente a la banda de esteroles sobre papel satinado e introducirle seguidamente en la columna de elución que deberá ser preparada de la forma siguiente:
– Introducir un tapón de algodón de vidrio en la extremidad del cuerpo intermedio de la columna, con un diámetro interno de 6 mm y ocupando el relleno una altura de 30 mm. Verter sobre este tapón el polvo obtenido por raspado de la placa, completando con silicagel fresca hasta llenar totalmente el tubo de 6 mm de diámetro.
– Verter por la boca de la columna, y en fracciones de 1 ml éter etílico, esperando para efectuar cada adición que la fracción precedente haya penetrado en la columna de sílice. Efectuar 6 adiciones, totalizando 6 ml. Recoger la solución en un matraz en forma de pera de 10 ml y eliminar el disolvente en evaporador rotatorio. El residuo sólido se disuelve en 250 µl de cloroformo. Esta solución es la que se inyectará en el cromatógrafo.
24(a).4.3 Cromatografía gaseosa de los esteroles.
24(a).4.3.1 Preparación de la columna. La preparación de la columna cromatográfica y su acondicionamiento se efectuará siguiendo las normas habituales en cromatografía gaseosa, debiendo ajustarse a las características que se indican en 24(a).2.7.
24(a).4.3.2 Condiciones de trabajo. No es posible indicar unas condiciones estrictas de trabajo que dependerán, fundamentalmente, del comportamiento de la columna y, en menor grado, de las características del aparato.
A título de orientación, se indican a continuación las condiciones de trabajo alrededor de las cuales se encuentran los resultados óptimos en la mayoría de los equipos utilizados:
‒ Temperatura de columna: 240-250 ºC.
‒ Temperatura del inyector: 290-300 ºC.
‒ Temperatura del detector: 270 ºC.
(Si ésta es independiente):
‒ Flujo del gas portador (N2): 30-40 ml/min.
La marcha de la operación y el comportamiento de la columna pueden considerarse satisfactorios si se registra para el β-sitosterol un pico simétrico, sin aparición de «colas» y que la resolución del par campesterol-estigmasterol no sea inferior a 0,80.
Normalmente el máximo del pico del β-sitosterol se suele registrar a los treinta minutos del disolvente, pero pueden producirse algunas variaciones sin que afecte a la eficacia de la operación.
24(a).4.3.3 Inyección de la muestra. De la solución en cloroformo de los esteroles separados por cromatografía en capa fina, se inyecta de uno a dos µl aproximadamente, que deberá contener de 10 a 15 µg para el componente mayoritario, 0,15-0,20 µg para el componente minoritario.
Si el registro obtenido fuera demasiado pobre y no se pudiese aumentar el tamaño de los picos, hasta una magnitud suficiente, disminuyendo la atenuación de la señal del aparato, se aumentará el volumen de solución inyectada. Para fines cuantitativos el registro si es para aceites de semillas se podrá hacer con la misma atenuación para todos los picos o introducir atenuación 1/2 para el pico del β-sitosterol. Si el análisis es para aceite de oliva, se efectuará con una atenuación de 1/2 para el pico del β-sitosterol, si es posible cuantificar el colesterol para un contenido mínimo de 0,5 por 100, de no ser posible se atenuará en 1/4 el pico del β-sitosterol.
24(a).4.3.4 Interpretación de los registros. Se utilizarán como criterio de identificación los tiempos de retención relativos al β-sitosterol, cuyos valores normales, en las condiciones de trabajo dadas, son los siguientes:
‒ Colesterol: 0,60.
‒ Brasicosterol: 0,68.
‒ Composterol: 0,80.
‒ Estigmasterol: 0,87.
‒ β-sitosterol: 1,00.
‒ Δ 7-sigmasterol: 1,16.
En el caso de que se quiera efectuar una comprobación directa de los tiempos de retención, preparar una solución de esteroles patrones en cloroformo al 1 por 100 respecto a cada uno.
Si no se dispone de patrones de calidad adecuada, se podrá obtener para fines cualitativos una solución patrón, extrayendo los esteroles de una mezcla en pesos iguales de aceite de colza y manteca de cacao, operando como en el procedimiento descrito. A la solución de esteroles recuperada en la placa, adicionar 1 mg de colesterol.
24(a).5. Cálculo.
Para la cuantificación de los registros con fines de caracterización de la muestra, se empleará el método de normalización interna, tomando en cuenta exclusivamente los cinco primeros esteroles mencionados en el cuadro anterior, y que estuviesen presentes en el problema.
Ajustándose estrictamente a las condiciones operativas señaladas, el factor de respuesta se considerará igual a la unidad para todos los esteroles.
En caso de que se disponga de integrador electrónico, las cuantificaciones se habrán multiplicado los tiempos de retención corregidos por la altura de cada pico, considerándose este producto como un estimado del área.
El contenido, por 100, de cada esterol en la fracción vendrá dado por la expresión siguiente:

La cuantificación se efectuará sólo para los cinco primeros esteroles si los contiene (para aceites de oliva), y de todos los esteroles para aceites de semillas.
24(a).6 Observaciones.
24(a).6.1 La resolución se calcula por la fórmula usual:

Siendo:
d2 y d1 = distancias, en el registro, del máximo de los picos al frente de salida del disolvente.
b2 y b1 = bases de los picos.
24(a).6.2 En el caso de analizar grasas de origen animal, especialmente grasas de leche, muy rica en colesterol y con el fin de identificar esteroles de origen vegetal, se recomienda inyectar una cantidad mayor de esteroles, sin sobrepasar la cifra de 20-30 µg de colesterol; pudiéndose efectuar, una vez registrado el pico del colesterol, un aumento de sensibilidad, disminuyendo la atención en la relación 4:1.
24(a).6.3 Para los análisis de aceite de oliva se utilizará como fase fija la SE-30 para el relleno de la columna.
24(a).7 Referencias bibliográficas.
1. Instituto Español de Normalización. UNE 55.019.
2. Determinación del insaponificable UNE 55.004.
Columna de elución
Cotas en milímetros
54. Actividad de adsorbentes para cromatografía en columna
54.1 Principio.
Se fija la actividad del adsorbente que, según su comportamiento frente a seis colorantes orgánicos. La actividad se expresa en cinco grados, designados en orden creciente del I al V. En los grados intermedios se establecen para cada una de los subgrados: 1 y 2.
Este método es aplicable a los adsorbentes gel de sílice y alúmina, en los diferentes tipos utilizados para cromatografía en columna, pudiéndose aplicar a otros adsorbentes de características físico-químicas análogas.
54.2 Material y aparatos.
Columnas de cromatografía, de 100 mm de longitud y 15 mm de diámetro interior, provistas de llave esmerilada, con orificio de paso de 1.5 a 2 mm v placa filtrante de vidrio de porosidad número 2, correspondiente a un tamaño medio de poro de 40 a 90μm.
54.3 Reactivos.
54.3.1 Azobenceno (a).
54.3.2 p-metoxiazobenceno (b).
54.3.3 Amarillo Sudan (Sudan I) (Benceno-azo-β-naftol) (c).
54.3.4 Rojo Sudan (Sudan II) (xileno-azo-naftol) (d).
54.3.5 p-Aminoazobenceno (e).
54.3.6 p-Oxiazobenceno (f).
54.3.7 Benceno, exento de tiofeno.
54.3.8 Hexano.
54.4 Procedimiento.
54.4.1 Preparación de la columna. El adsorbente que se ha de ensayar, se introduce en la columna cromatográfica, procurando conseguir una distribución homogénea, formando una capa de 50 mm de altura, colocando, previamente, un disco de papel de filtro en la parte inferior, y una vez empaquetado el adsorbente, se coloca encima otro disco de papel de filtro.
54.4.2 Preparación de las disoluciones de colorantes para los ensayos. Se preparan soluciones pareadas de los colorantes indicados en el apartado 50.3 pesando 2 mg de cada colorante que se disuelven en una mezcla de 2 ml de benceno y 8 ml de hexano.
Las disoluciones que constituirán una serie completa para medir la gama de actividad de la I a la V, serán las siguientes:
Solución 1: 2 mg de a y 2 mg de b.
Solución 2: 2 mg de b y 2 mg de c.
Solución 3: 2 mg de c y 2 mg de d.
Solución 4: 2 mg de d y 2 mg de e.
Solución 5: 2 mg de e y 2 mg de f.
54.4.3 Determinación. Verter sobre la columna preparada como se indica en (54.4.1), 10 ml de la disolución adecuada para la actividad que se prevé para el producto ensayado, abrir la llave, dejando pasar la disolución hasta que el nivel líquido queda justamente a la altura del relleno de la columna.
Verter el líquido de desarrollo constituido por una mezcla de 4 ml de benceno y 16 ml de hexano. La velocidad de salida se regula con la llave, manteniendo un flujo de 1 ml/minuto, aproximadamente, equivalente a la caída de 1 gota cada 2 segundos. Recoger el líquido en un matraz cónico de boca estrecha.
54.4.4 Expresión de los resultados. Terminada la salida del líquido de desarrollo, se observa la situación de los colorantes. En el cuadro que se incluye a continuación se muestra un esquema de las posiciones de los pares de colores con las distintas soluciones de ensayo, en función de la actividad del adsorbente. Buscando la posición que se corresponda con la observada en el ensayo, en la columna correspondiente encontraremos el grado de actividad del adsorbente ensayado.
En el caso de que el adsorbente se manifiesta con una actividad superior o inferior a la prevista, se repetirá el ensayo utilizando la solución de colorantes que se considere adecuada.
Si al utilizar la solución 1, los dos colorantes quedasen fuertemente retenidos en la parte superior de la columna, el adsorbente ensayado se comportará con una actividad mayor que I.
Análogamente, una elución completa para los colorantes de la solución número 5, indicará una actividad inferior a V.
| Grado de actividad: | I | II | III | IV | V | |||
|---|---|---|---|---|---|---|---|---|
| II1 | II2 | III1 | III2 | IV1 | IV2 | 5 | ||
| Número de la solución: | 1 | I | 2 | 2 | 3 | 3 | 4 | |
| Posición en la columna: | ||||||||
| Parte superior: | a | b | c | c | d | d | e | f |
| Parte inferior: | a | |||||||
| En el filtrado: | a | b | c | |||||
54.6 Referencias bibliográficas.
1. Instituto Español de Normalización. UNE 55.129.
55. Ceras en aceites de girasol
55.1 Principio.
Precipitación de los productos insolubles en acetona, ceras y gran parte de fosfátidos. Eliminación por lavados sucesivos de los triglicéridos arrastrados y separación cuantitativa de las ceras por cromatografía en columna de alúmina.
El método es aplicable a aceites de girasol brutos y refinados.
55.2 Material y aparatos.
55.2.1 Estufa de desecación, con regulador de temperatura, pudiéndose calentar hasta 150 ºC.
55.2.2 Erlenmeyers de 250 y 100 ml, con boca esmerilada.
55.2.3 Embudo de vidrio, forma alemana, de 6 cm Ø.
55.2.4 Pesafiltro de 3 cm de diámetro y 7 cm de altura.
55.2.5 Papel de filtro «Albet 240» o similar, en discos de 8 cm de diámetro.
55.2.6 Cápsulas de vidrio, fondo plano, de 5-6 cm de diámetro.
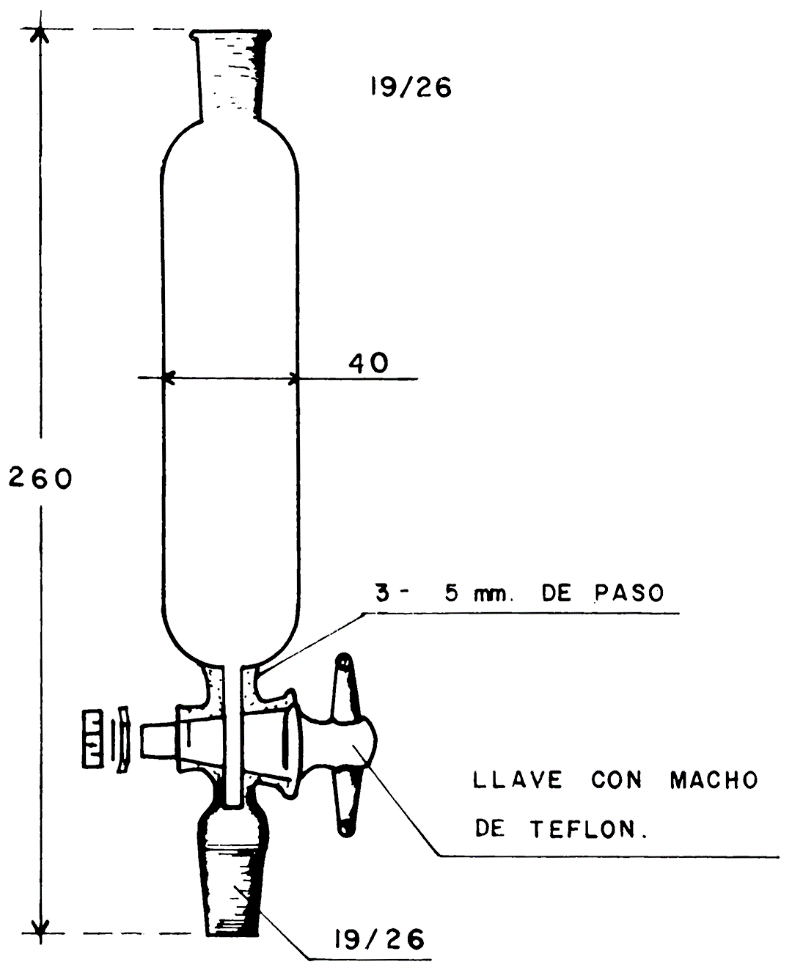
55.2.7 Columna cromatográfica, con camisa de calefacción y dimensiones indicadas en la figura I, y depósito de disolvente para la alimentación de la columna, representado en la figura 2.
55.2.8 Termostato de corriente, para regulación de temperatura de dispositivos exteriores, destinados a su utilización con la columna cromatográfica (55.2.7). Deberá tener las especificaciones mínimas siguientes:
Temperatura máxima alcanzable: 60 ºC.
Precisión en la regulación de la temperatura: ± 0,05.
Potencia de calefacción: 750 W.
Precisión máxima de la bomba de circulación: 1,5 m columna de agua.
Dispositivo de regulación adecuado a las especificaciones anteriores.
55.2.9 Aparato de destilación, totalmente de vidrio, con matraz fondo plano, de 500 ml, columna «Vigreux» de 15 cm y refrigerante «Liebig» de 30 cm; el destilado se recoge en un kitasato de 500 ml.
55.2.10 Bloque metálico de calefacción, capaz de alcanzar una temperatura de 100 ºC y con regulación de temperatura con una precisión de ± 1 ºC.
55.2.11 Material necesario para el ensayo de la alúmina (según método oficial número 54).
55.3 Reactivos.
55.3.1 Acetona anhidra, calidad reactivo para análisis.
55.3.2 Benceno, calidad reactivo para análisis, exento de tiofeno.
55.3.3 Alúmina para cromatografía en columna, que debe ajustarse a las especificaciones siguientes:
En suspensión acuosa al 5 por 100 el pH será de 7.0 ± 0,5.
Actividad IV según método oficial número 54, determinada y ajustada según se indica:
El resultado del análisis granulométrico del producto debe ser tal que su contenido en partículas con un grosor igual o inferior a 63 µm no llegue a 10 por 100; el de partículas con un grosor mayor de 250 µm no llegue a 0.1 por 100; y la media estadística de tamaños para la totalidad de las partículas sea de 130µm ± 5 (55.6.2).
55.3.4 Reactivas para la determinación de la actividad de la alúmina. Según método oficial número 54.
55.4 Procedimiento.
55.4.1 Determinación de la actividad de la alúmina. Según método oficial número 54.
55.4.2 Preparación de la columna cromatográfica.
La cantidad necesaria de alúmina que es de 16,5 g ± 0,5 g se introduce en la columna, pudiéndose utilizar cualquiera de los procedimientos usuales, operando ya sea en seco o en húmedo y siempre que se consiga una distribución homogénea del relleno.
El procedimiento recomendado es el siguiente: La cantidad pesada de alúmina se introduce en una ampolla de extracción de 125 ml agregando 50 ml de benceno con el que se llena, también, la columna hasta ocupar, aproximadamente, 2/3 de su capacidad. Se coloca la ampolla encima de la columna, introduciendo en ella un tubo de salida. Se abre la llave de la ampolla dejando salir la papilla de alúmina, que caerá en el interior de la columna con el disolvente; al mismo tiempo, se abre la llave de la columna regulando la salida del benceno de forma que el nivel del líquido en el interior de la columna se mantenga prácticamente constante, procurando, más bien, que el nivel suba ligeramente en lugar de disminuir. Cuando toda la papilla de alúmina haya pasado a la columna, se arrastran las últimas porciones adheridas a las paredes, enjuagando el recipiente con un poco de benceno.
Se mantiene abierta la llave de la columna, dejando salir lentamente el disolvente hasta que el nivel se haya situado a la altura del relleno de la columna. La columna queda así lista para su empleo.
55.4.3 Preparación de la muestra. La muestra deberá estar seca y perfectamente homogeneizada en el momento de efectuar la pesada.
En el caso de contener pequeñas cantidades de humedad, o no estuviese transparente, se calentará en estufa a unos 50 -55 ºC, para asegurar la disolución de las ceras que pudieran haber precipitado, agitando para asegurar la homogeneización y filtrando en el interior de la estufa.
Estas operaciones deberán realizarse en el tiempo más breve posible.
En el caso de que el agua no quedara eliminada en esta filtración, se deberá desecar en estufa de vacío hasta constancia de peso en la cifra de los miligramos.
55.4.4 Separación de la materia insoluble en acetona. En un erlenmeyer de 250 ml, pesar, con exactitud del centigramo, la cantidad necesaria de muestra que debe tener un contenido de ceras de 8 mg a 15 mg. Normalmente, en los aceites brutos deberán pesarse de 10 a 15 g de muestra; en aceites refinados e invernadas sería necesario operar con unos 50 g.
Tapar el erlenmeyer e introducirlo en un frigorífico a una temperatura de 0-5 ºC, manteniéndolo allí de treinta a cuarenta minutos.
Transcurrido este tiempo, se saca el erlenmeyer, agregando 100 ml de acetona anhidra, enfriada previamente a una temperatura de 0 -5 ºC, para cantidades de muestra no superior a 15 g; para muestras comprendidas entre 16 g y 50 g, se agregarán 150 ml de acetona. Colocar el tapón y agitar fuertemente hasta observar la formación de grumos que quedan en suspensión. Dejar en reposo, en frigorífico, a unos 5C º durante unos treinta minutos, debiéndose depositar en el fondo el precipitado de carácter algodonoso, quedando la solución acetónica limpia y transparente; si se observa la disolución turbia o el sedimento no tuviese el aspecto algodonoso anteriormente aludido, la operación no estaría bien realizada, debiendo repetirse nuevamente.
Filtrar la disolución por papel de filtro, previamente desecado en un pesafiltro y en estufa a 105 ºC y enfriado en desecador y pesado, cuidando de pasar todo el precipitado al filtro, ayudándose de acetona anhidra, previamente enfriada a una temperatura inferior a 5 ºC como se dijo anteriormente. Lavar el filtro y su contenido con la misma acetona fría hasta eliminación total de la grasa, cuidando de emplear en todas estas operaciones la menor cantidad posible de acetona, no debiendo, sobrepasar el volumen total utilizado de unos 100 ml.
El filtro con el precipitado, una vez lavado e introducido en el pasafiltro, se lleva a una estufa regulada a 70 ºC, manteniéndolo dos horas. Se deja enfriar en desecador y se pesa.
El peso obtenido constituye el denominado insoluble total en acetona, constituido, fundamentalmente, por las ceras y los fosfátidos del aceite.
55.4.5 Cromatografía. El filtro conservado en el pasafiltro y conteniendo el precipitado se coloca en un embudo, que se introduce en la boca de un erlenmeyer de 100 ml disolviendo el precipitado en benceno caliente, que se vierte en pequeñas porciones en el embudo (55.6.3), y lavando convenientemente el filtro con benceno caliente.
La disolución de benceno se pasa a una cápsula de vidrio, concentrándose en el bloque de calefacción o al baño María, operando en vitrina con un buen tiro hasta reducir el volumen a unos 10 ml (55.6.4).
La disolución se vierte sobre la columna de alúmina, preparada según 55.4.2 y calentada a 40 ºC, lavando la capsulita dos veces con 2 ml de benceno cada vez, que se vierten, también, en la columna.
Una vez que la disolución de benceno ha penetrado en la columna, añadir 250 ml de benceno en el depósito de alimentación, regulándose la salida de la columna cromatográfica con la llave situada en la parte inferior, a una velocidad aproximada de una gota por segundo (unos 2 ml/min), regulándose también la salida del depósito del disolvente (figura 2), de manera que el nivel de benceno se mantenga por encima del nivel de la alúmina.
Se continúa el desarrollo hasta que se consuma la totalidad del benceno.
El eluato recogido se destila hasta reducir su volumen a unos 20-25 ml, que se trasvasan a una cápsula de vidrio (55.2.6), seca y tarada previamente, lavado con dos o tres pequeñas porciones de benceno para hacer el trasvase cuantitativo.
Se evapora el líquido de la cápsula hasta sequedad, calentando para ello en bloque de calefacción, regulado a 75 -80 ºC y bajo vitrina (55.6.3), sin que llegue a hervir el benceno.
Se deja enfriar en desecador y se pesa (55.6.5).
55.5 Cálculos.

P1 = peso, en gramos, de la cápsula con el residuo.
P0 = peso, en gramos, de la cápsula vacía y seca.
M = peso tomado de muestra, en gramos.
55.6 Observaciones.
55.6.1 La tapa deberá ser de un material no atacable por la acetona ni por el benceno.
55.6.2 Los valores que se señalan en el apartado 55.3.3, para el grosor de las partículas de la alúmina que se recomienda utilizar, se corresponden con las aberturas de mallas de los tamices que se indican a continuación:
|
Luz de malla ‒ mm |
Numeración |
|---|---|
| 0,063 | Tamiz 0,063 |
| 0,25 | Tamiz 0,25 |
55.6.3 Dado el carácter tóxico del benceno, debe evitarse la respiración de sus vapores, efectuándose todas las operaciones en vitrina con un buen tiro, manteniendo la compuerta de cierre lo más baja posible.
55.6.4 Si se dispone de evaporador rotatorio, la concentración de la disolución puede hacerse con más rapidez y comodidad en el mismo erlenmeyer, sin tener que trasvasar a la cápsula.
55.6.5 Las ceras, una vez desecadas, deben tener un color blanco limpio. Una coloración generalmente amarillenta o ambarina indica haberse realizado una purificación incompleta, falseando los resultados.
55.6.6 Es la máxima importancia la calidad de la acetona, cuyo contenido de agua es crítico para la marcha normal del proceso de insolubilización. Por este motivo, es conveniente someter la acetona anhidra comercial, calidad relativo análisis, a un proceso previo de deshidratación, habiendo dado buen resultado el tratamiento• con cloruro cálcico escoriforme, que se deja en contacto con la acetona durante cuatro o cinco horas como mínimo, destilando la cantidad necesaria inmediatamente antes de su utilización, empleando una columna «Vigreux» de 30 cm.
55.7 Referencias bibliográficas.
1. Instituto Español de Normalización. UNE 55.115.
2. Instituto Español de Normalización. UNE 55.129.
56. Tocoferoles
56.1 Principio.
1. Extracción de la materia insaponificable de la grasa.
2. Aislamiento de la fracción de tocoferoles por cromatografía en capa fina.
3. Separación y cuantificación de los componentes de la fracción de tocoferoles por colorimetría o cromatografía gaseosa.
56.2 Material y aparatos.
56.2.1 Matraces de fondo redondo de 50 ml y 100 ml.
56.2.2 Matraces en forma de corazón de 15 ml.
56.2.3 Tubos de reflujo de 1 ml para adaptar a los matraces (56.2.1) o refrigerante de reflujo.
56.2.4 Ampollas de decantación de 125 ml.
56.2.5 Microjeringuillas de 100 µl.
56.2.6 Placas de vidrio para cromatografía en capa fina de 20 cm x 20 cm, con capa de gel de sílice de 0,25 mm de espesor.
56.2.7 Contraplacas de 20 cm x 16 cm.
56.2.8 Cubeta de desarrollo en vidrio para cromatografía en capa fina, con tapa de vidrio.
56.2.9 Pulverizador para líquidos de revelado.
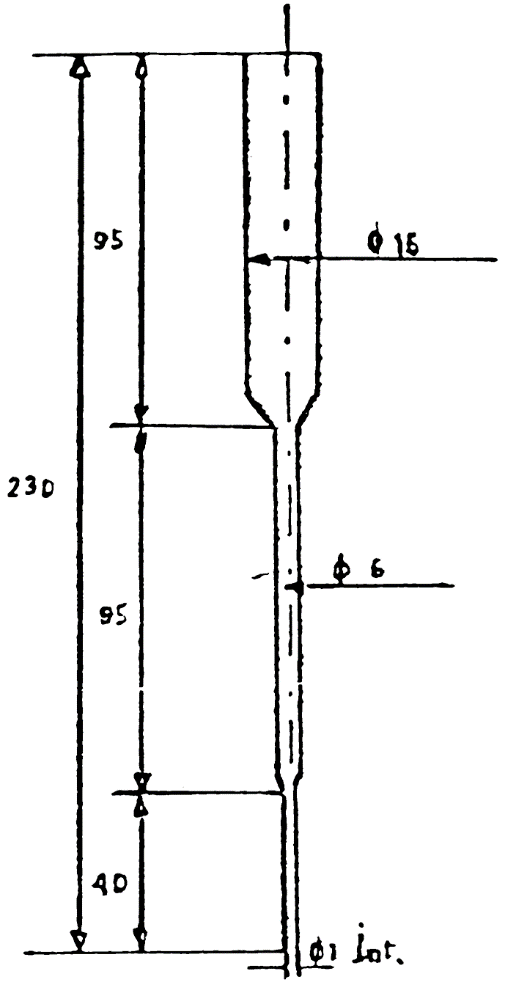
56.2.10 Columna de elución, de vidrio, diámetro interior 6 mm, con un tubo superior de 16 mm de diámetro y 95 mm de longitud, con un tubo capilar inferior de 40 mm de longitud (figura 1).
56.2.11 Evaporador rotatorio o bario de agua hirviendo.
56.2.12 Estufa regulada a 110 ± 2 ºC
56.2.13 Pinzas para fijar las contra placas.
56.2.14 Espectrofotómetro para lectura en el espectro visible.
56.2.15 Cubetas de 1,00 cm ± 0,01 cm de espesor.
56.2.16 Matraces de fondo redondo de 10 ml con tapón esmerilado.
56.2.17 Microjeringuillas de 1 µl.
56.2.18 Apartado de cromatografía gaseosa con detector de ionización de llama y registrador.
56.2.19 Columna de vidrio de unos 2 m de longitud, diámetro interno 2,2-2,5 mm, con relleno de 3 al 5 por 100 de metilsilicona (SE-30), o fenilmetilsilicona (OV-17), sobre tierra de diatomas, con granulometría de 80-100 mallas.
56.2.20 Columna capilar de vidrio, longitud 20 m, diámetro interno 0,4 mm de metilsilicona (SE-30), o fenilmetilsilicona (0V-17).
56.3 Reactivos.
56.3.1 N-Hexano para cromatografía.
56.3.2 N-Heptano para cromatografía.
56.3.3 Benceno anhidro (contenido en agua máximo 0,01 por 100).
56.3.4 Etanol absoluto exento de aldehídos, de calidad analítica.
56.3.5 Éter etílico, exento de peróxidos y de residuo.
56.3.6 Líquido de desarrollo: Hexano-eter 7:3 (V/V).
56.3.7 Líquido de elución: Heptano-etanol 2:1 (V/V).
56.3.8 Pirogalol (1, 2, 3, Bencenotriol), solución etanólica de 5 g/100 ml, preparada inmediatamente antes del empleo.
56.3.9 Hidróxido de potasio, solución acuosa de 1.000 g/l.
56.3.10 Solución de cloruro de hierro (111): Disolver 200 g de (FeCl, 6H20), de calidad analítica en 100 ml de etanol absoluto.
Fig. 1 -Columna
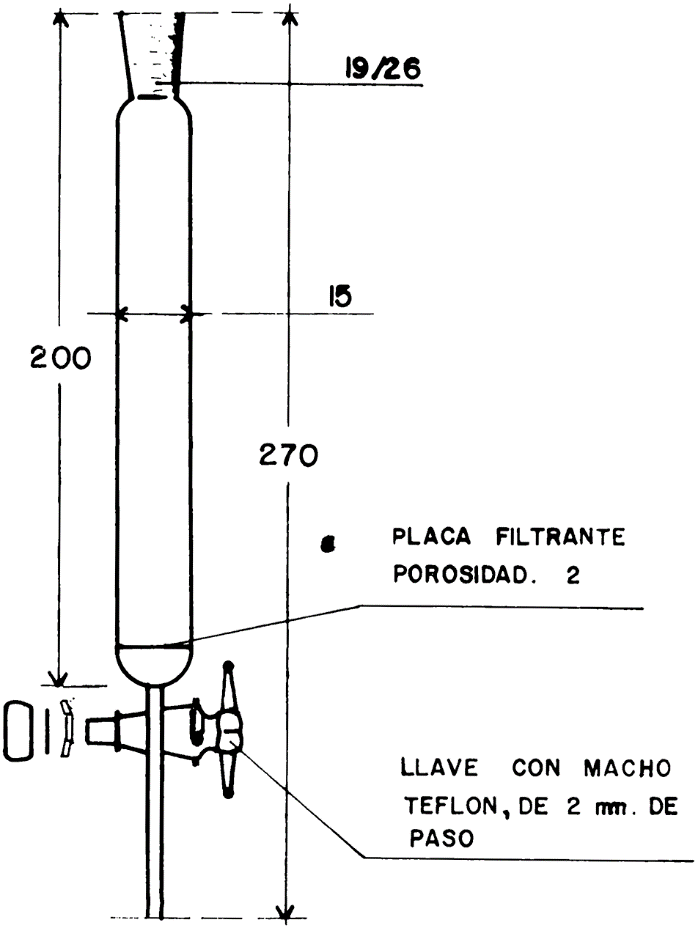
Fig. 2 - Depósito
Cotas en milímetros
La solución se debe preparar inmediatamente antes del empleo.
56.3.11 Solución de referencia de tocoferoles: Disolver 200 mg de una mezcla en partes iguales de los tres tocoferoles α, β y δ en 10 ml de heptano. Puede sustituirse esta solución por otra obtenida a partir de mezcla de aceite de soja y girasol, siguiendo el procedimiento (56.4.1) (56.4.2).
56.3.12 Solución de fenolftaleína en etanol absoluto: 10 g/l.
56.3.13 Nitrógeno conteniendo menos de 5 mg de oxígeno por kg.
56.3.14 Disolución etálica de 2,2'-dipiridilo. Disolver 200 mg de 2,2' dipiridilo en 100 ml de etanol absoluto.
56.3.15 Piridina anhidra, de calidad analítica, recientemente destilada sobre hidróxido de sodio u óxido de bario.
56.3.16 Hexametildisilazano puro.
56.3.17 Dimetilclorosilano puro.
56.3.18 Escualano puro.
56.3.19 Solución de escualano en n-heptano, 100 mg /100 ml. 56.3.20 d-1α tocoferol, pureza mínima 99 por 100.
56.4 Procedimiento.
56.4.1 Extracción del insaponificable total. Pesar, con presión de 0,005 g, 1 g de muestra en un matraz de 50 ml. Añadir 4 ml de la solución etanólica de pirogalol. Adaptar el refrigerante de reflujo y llevar a ebullición. Al empezar la ebullición añadir 1 ml de la solución de hidróxido de potasio. Dejar en ebullición durante tres minutos. Retirar el matraz y refrigerarlo bajo una corriente de agua, añadir 25 ml de agua destilada.
Trasvasar cuantitativamente a una ampolla de decantación. Enjuagar el matraz con 40 ml de éter etílico, añadirlo a la ampolla y efectuar una primera decantación. Efectuar otras dos extracciones de la fase acuosa utilizando cada vez 25 ml de éter etílico. A cada extracción agitar suavemente la ampolla de decantación, girando sobre sí misma, para evitar la formación de emulsiones, dejar reposar para separar la fase acuosa.
Reunir los tres extractos en la ampolla de decantación y lavar con porciones de 20 ml de agua, agitando enérgicamente hasta que el líquido de lavado no vire a rosa por adición de una gota de la solución de fenolfetalina. Trasvasar la fase etérea a un matraz de 100 ml y evaporar el éter etílico por destilación en un evaporador rotatorio o calentando sobre un baño de agua hirviendo. Para secar el residuo, añadir 1 ml de etanol y 4 ml de benceno y evaporar en corriente de nitrógeno por destilación en un evaporador rotatorio o calentando sobre baño de agua hirviendo. Repetir la adición de etanol y benceno y evaporar estos disolventes.
Disolver el residuo en 1 ml de hexano y transferir a un matraz en forma de corazón, utilizando la menos cantidad posible de hexano para los lavados. Evaporar totalmente el hexeno al vacío o con comente de nitrógeno. Eliminar el aire del matraz con ayuda de una comente de nitrógeno y añadir exactamente 1 ml de heptano. Efectuar inmediatamente la cromatografía en capa fina de la solución obtenida.
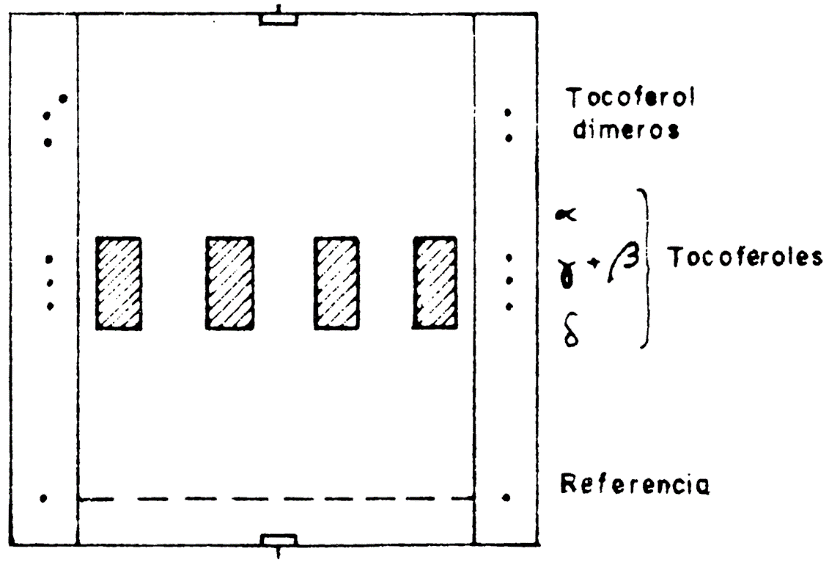
56.4.2 Cromatografía en una capa fina. Todo el procedimiento descrito a continuación deberá efectuarse en ausencia de luz natural, en una habitación alumbrada por una lámpara eléctrica recubierta con papel rojo claro. Saturar la cubeta cromatográfica con el líquido de desarrollo (56.3.6) y cubrir las caras Internas de la cubeta con papel de filtro. Sobre una placa de cromatografía en capa previamente activada, calentando treinta minutos en una estufa a 110 ºC y con la ayuda de una microjeringa graduada, depositar 1μl de la solución de tocoferoles de referencia (56.3.11), a 2 cm de cada borde lateral y a 1 cm del borde inferior. A partir de 6 cm del borde derecho y sobre una banda de 2 cm de longitud, depositar con microjeringa en finas gotas lo más uniformemente posible la solución del insaponificable obtenida en 56.4.1.
La cantidad de insaponificable depositada estará comprendida entre 25 y 72µ 1 según la riqueza del aceite en tocoferoles; normalmente la cantidad depositada es de 50 µl. Esta cantidad deberá ser medida exactamente en el caso de una determinación cuantitativa. Si se pone varias muestras para analizar en la misma placa, deberán depositarse las bandas con un espacio de separación de 2 cm entre cada muestra.
Introducir inmediatamente la placa en la cubeta en ausencia de luz, hasta que el frente del líquido de desarrollo diste 1 cm del borde superior de la placa. Sacar la placa y cubrirla inmediatamente con la contraplaca sujetándola con ayuda de dos pinzas, dejando sin cubrir dos bandas a la derecha y a la izquierda de la placa sin cubrir. Pulverizar inmediatamente las dos bandas laterales descubiertas, poniendo en el pulverizador una mezcla en volúmenes iguales de las soluciones de cloruro de hierro (III) y de 2,2' dipirilo (56.3.14). Conservar la placa en la oscuridad durante cinco o diez minutos. La aparición de manchas de color rosa oscuro indica la posición de-los tocoferoles de la solución de referencia.
Quitar la contraplaca y señalar los rectángulos de las bandas de 2 cm de los tocoferoles, a las alturas indicadas, por las manchas reveladas de los tocoferoles de referencia. Rascar la sílice contenida en cada rectángulo marcado, siguiendo las precauciones habituales para que las repercusiones sean cuantitativas.
Con la ayuda de un embudo pequeño introducir la sílice de cada rectángulo en su columna (56.2.10). Añadir pequeñas porciones y facilitar el empaquetamiento con ligeros golpes sobre las paredes de la columna.
Llevar la columna sobre un matraz en forma de corazón. Pasar 5-6 ml del disolvente de elución (56.3.7). Evaporar los disolventes al vacío y proceder inmediatamente a la evacuación de los tocoferoles en el residuo, por uno de los métodos de cuantificación.
56.4.3 Colorimetría Todas las operaciones siguientes deberán efectuarse en cámara y con luz roja.
En cada matraz conteniendo la fracción tocoferólica obtenida en 56.4.2, añadir exactamente 3,6 ml de etanol, 0,2 ml de la solución de cloruro de hierro III (56.3.10). Mezclar por ligera rotación del matraz y dejar reposar durante diez minutos.
Efectuar un ensayo en blanco en las mismas condiciones.
Trasvasar las soluciones a las cubetas (56.2.15) y medir las absorbancias a 520 nm de las muestras y del ensayo en blanco, con relación al etanol. El ensayo en blanco debe tener una absorbancia inferior a 0,05.
Este método es aplicable a todos los aceites, incluidos los de oliva virgen y no se permite la separación del γ y β y β tocoferol, ni de los tocotrienios correspondientes.
56.4.4 Cromatografía gaseosa.
56.4.4.1 Preparación de los sililéteres.
En un matraz (56.2.16) añadir 0,5 ml de piridina (56.3.15), 0,45 ml de hexamerildisilazano (56.5.16) y 0,3 ml de dimetilclorosilano.
Tapar y dejar reposar quince minutos hasta la completa decantación del precipitado blanco.
Del líquido superior claro, tomar I00µ 1 con microjeringa e introducirlo en un matraz que contiene la fracción de tocoferoles en 56.4.2. La mezcla de la solución se facilita inclinando el matraz y haciendo girar sobre sí mismo. Esperar cinco minutos antes de proceder a la cromatografía gaseosa.
56.4.4.2 Condiciones de trabajo.
Para las columnas dadas en 56.2.19 serán:
Temperatura de la columna: 240 ºC.
Temperatura del inyector: 250 ºC.
Temperatura del detector: 250 ºC.
Flujo del gas portador: 0,8 atmósferas.
Inyectar 0.5-1 μl de la solución silanizada que, según la respuesta cromatográfica puede ser concentrada por evaporación en corriente de nitrógeno.
En estas condiciones y utilizando la columna (SE 30), los tiempos de retención relativos al α-tocoferol son:
α-tocoferol 1,00.
γ + β tocoferol 0,71.
δ-tocoferol 0,55.
56.4.4.3 Para columnas capilares (56.2.20), las condiciones cromatográficas serán:
Temperatura de la columna: 240 ºC.
Temperatura del inyector: 250 ºC.
Temperatura del detector: 250 ºC.
Gas portador caudal de entrada: 2 ml/min; caudal de columna: 60 ml/min.
Evaporar los relativos en corriente de nitrógeno de los sililéteres obtenidos según 56.4.1. Disolver el residuo en 1 ml de éter etílico e inyectar en el cromatógrafo 1 µ litro.
En estas condiciones, los tiempos de retención reducidos para el α-tocoferol serán los siguientes:
| OV-17 | SE-30 | |
|---|---|---|
| α-tocoferol | 1,00 | 1,00 |
| β-tocoferol | 0,64 | 0,68 |
| γ-tocoferol | 0,66 | 0,70 |
| δ-tocoferol | 0,50 | 0,54 |
| α-tocotrienol | 1,71 | 1,31 |
| β-tocotrienol | I,09 | 0,89 |
| γ-tocotrienol | 1,13 | 0,92 |
| δ-tocotrienol | 0,86 | 0,70 |
56.4.4.4 Determinación cuantitativa. Determinación de la curva patrón. En cuatro matraces (56.2.16), pesar exactamente α-tocoferol y escualano en las proporciones: escualano/α-tocoferol. 3/1, 2/1. 1/1, 0,5/1.
Preparar los sililéteres como se indica en 56.4.4.1 y proceder al análisis cromatográfico. Trazar la curva poniendo en abscisas la relación: pesos de esculeno/pesos de α-tocoferol y en ordenadas la relación, área del pico de escualano/área del pico de α-tocoferol. Esta curva debe ser una recta que pase por el origen. A partir de esta curva, calcular la relación:

56.4.4.5 Introducción del patrón interno. En el matraz conteniendo la fracción de tocoferoles obtenida en 56.4.2, añadir 1 ml, exactamente medido, de la solución de escualano. Evaporar el disolvente al vacío y proceder inmediatamente a la silanización según 56.4.4.41.
Operar exactamente como en 56.4.4.2.
En estas condiciones y utilizando la columna SE-30, los tiempos de retención relativos al escualano son los siguientes:
α-tocoferol: 3,36.
γ + α-tocoferol: 2,40.
δ-tocoferol: 1,85.
Escualano: 1,00.
56.4.4.6 En el caso de efectuar la cromatografía con columna capilar, introducir el patrón interno como en 56.4.4.5, y efectuar la cromatografía como se indica en 56.4.4.3.
56.5 Operaciones.
56.5.1 Colorimetría. En el contenido en tocoferol expresado en mg/100 g viene dado por la siguiente fórmula:

Siendo:
A = absorbancia de la disolución de muestra.
Ao = absorbancia del ensayo en blanco.
V = volumen, en ml, del heptano utilizado para disolver el insaponificable.
v = Volumen en μl de la solución de insaponificable depositada sobre la placa.
m = masa en gramos de la muestra.
F = factor espectrofotométrico diferente para cada tocoferol.
F α-tocoferol: 98.
F γ + β tocoferol: 90.
F δ tocoferol: 75.
56.5.2. Cromatografía gaseosa.
56.5.2.1 Utilizando el método de normalización interna los porcentajes de cada tocoferol vienen dados por la fórmula:

Siendo:
Ax = área del pico correspondiente a cada tocoferol.
A = suma de áreas de todos los picos de tocoferoles.
56.5.2.2 Utilizando patrón interno, el contenido en cada tocoferol expresado en mg/100 g vendrá dado por la fórmula:

Siendo:
V = Volumen en µl de la solución de insaponificable depositada sobre la placa.
m = masa en g de la muestra.
Ai = área de los picos de los tocoferoles.
As = área del pico correspondiente al escualano.
R = relación calculada en 56.4.4.4.
56.6 Observaciones.
56.6.1 En caso de necesitar preparar etanol absoluto exento de aldehídos, proceder como se indica. Todas las operaciones deben efectuarse con material de vidrio. Añadir 2 g de hidróxido de potasio y 1 g de permanganato de potasio a 1 1 de etanol absoluto comercial pobre en aldehídos (99, 5 por 100 mínimo de alcohol de melaza). Hervir a reflujo durante treinta minutos después de destilar. Deshidratar sobre sulfato de calcio anhidro. Destilar de nuevo y proteger de la humedad.
Figura 1
Cotas en milímetros
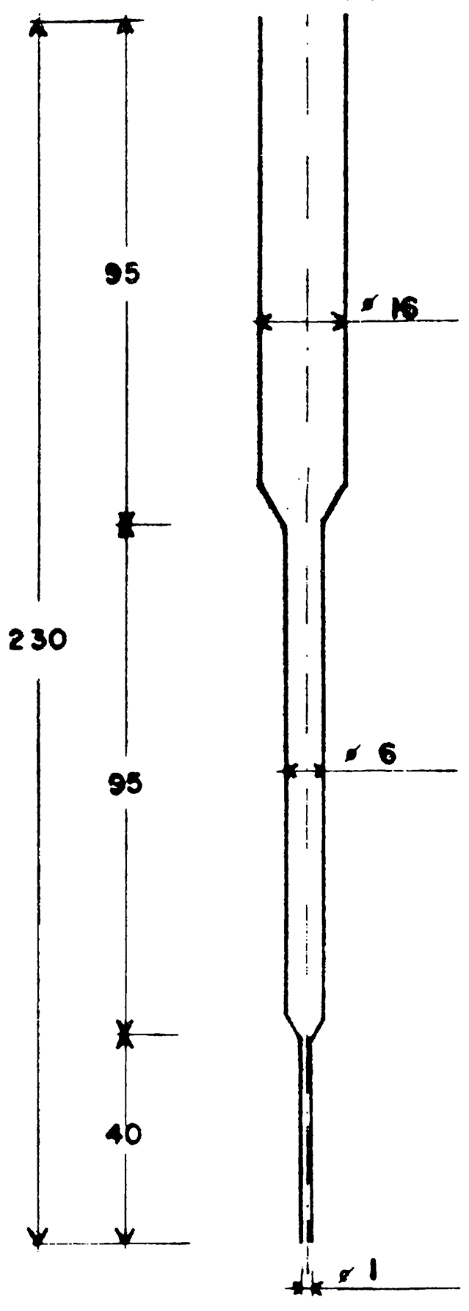
Figura 2
56.6.2. En caso de necesitar preparar éter etílico exento de peróxidos y de residuos, proceder como se indica:
En un frasco de vidrio oscuro agregar 130 g de tamiz molecular a 4 A en parlas, de 2 mm de diámetro por cada litro de éter etílico de calidad analítica. Agitar vigorosamente y dejar reposar durante doce horas en el mismo matraz cerrado. Conservar el éter etílico en estas condiciones.
Para eliminar los peróxidos, destacar el éter etílico, deshidratado, y pasarlo sobre una columna de alúmina básica, de 22 mm de diámetro y 600 mm de longitud, actividad 1, granulometría 0,063-0,200 mm, con 100 g de alúmina y un flujo de éter etílico alrededor de 2-3 ml/min. Recoger el elusto en un frasco de vidrio oscuro.
56.6.3 La solución etanólica de pirogalol puede ser sustituida por una solución etanólica de ácido ascórbico, obtenida por disolución de 0,3 g de ácido ascórbico en 4 ml de etanol preparada inmediatamente antes de utilizarse.
56.6.4 Durante los lavados con agua destilada, principalmente el primero, no se debe agitar enérgicamente para evitar la formación de emulsiones. En caso de formación de éstas, añadir algunos mililitros de una solución acuosa saturada de cloruro de sodio para romper la emulsión. La separación de las dos capas debe tener lugar inmediatamente.
56.7 Referencias bibliográficas.
1. International Union of Pure and applied Chemistry Standard, «Methods for the Analysis of Oils, Fats and Derivatines». 6.ª edición, primer suplemento.
57. Aceite semisecante en aceites de oliva y orujo.
57.1 Principio.
Cristalización fraccionada a baja temperatura de la muestra de aceite neutro en mezcla metanol-acetona. Separación de los triglicéridos más insaturados por filtración y posterior purificación por cromatografía en capa fina. Relación del contenido de ácido linoleico en esta fracción y en el aceite original.
El límite de detección se puede considerar entre el 1 al 5 por 100, dependiendo de la naturaleza de los aceites.
Este método es aplicable a aceites de oliva vírgenes, refinados y a sus mezclas, así como a los aceites de orujo brutos y refinados.
57.2 Material y aparatos.
57.2.1 Arcón de congelación, con unas dimensiones interiores aproximadas de 90x50 cm de base y 75 cm de profundidad, provisto de sistema de regulación de temperatura, que permita mantener en el interior, a una altura media del fondo, una temperatura de -22 ºC ± 1 ºC, durante un período mínimo de quince horas.
A unos 30 cm, aproximadamente, del extremo superior llevará una plataforma con las dimensiones suficientes para la colocación de los recipientes de cristalización y el material necesario para efectuar las operaciones de filtración, tal como se detalla en 57.4.2.
La tapa superior del arcón, coincidiendo con uno de los extremos de la plataforma, llevará un orificio para la introducción de un termómetro, graduado en décimas, de características adecuadas para verificar la temperatura de espacio en el que se van a realizar las operaciones de cristalización y filtración (57.6.5).
57.2.2 Equipo de cromatografía de capa fina.
57.2.3 Placas de vidrio de 20 x 20 cm.
57.2.4 Equipo de cromatografía gaseosa, preferiblemente provisto de integrador electrónico.
57.2.5 Tubos de ensayo con tapón esmerilado, de 2 cm de diámetro interior y 15 cm de altura. (Capacidad aproximada, 30 ml.)
57.2.6 Vasos de precipitado, forma alta, para su utilización como soporte de los tubos de ensayo o dispositivos similares.
57.2.7 Matraces de forma de corazón, con boca esmerilada y 50 ml de capacidad.
57.2.8 Soportes para los matraces de forma de corazón.
57.2.9 Embudo de vidrio, forma alemana de aproximadamente, 55 mm de diámetro exterior, vástago corto, con 4 mm, aproximadamente, de diámetro interior.
57.2.10 Vidrio de reloj de 58 mm de diámetro, aproximadamente.
57.2.11 Cubeta de vidrio con tapa esmerilada, para revelado de placas.
57.2.12 Papel de filtro Albet número 242, u otro equivalente, de 90 mm de diámetro.
57.2.13 Matraz de metilación redondo, fondo plano, de unos 50 ml de capacidad con boca esmerilada.
57.2.14 Varilla de reflujo adaptable al matraz anterior, para su utilización como refrigerante.
57.2.15 Ampolla de extracción de unos 500 ml de capacidad.
57.2.16 Probeta graduada, con tapón esmerilado de 25 ml.
57.2.17 Matraz redondo, fondo plano, de unos 250 ml de capacidad.
57.3 Reactivos.
57.3.1 Metanol R.A. con un contenido máximo de agua de 0,05 por 100.
57.3.2 Acetona R.A. con un contenido máximo de agua de 0,02 por 100.
57.3.3 Hexano.
57.3.4 Sílice gel Merck tipo 60 o similar para C.C.F.
57.3.5 Disolución de metilato de sodio 0,2N, en metanol absoluto.
57.3.6 Disolución de ClH gaseoso seco al 4 por 100, en metanol absoluto.
57.3.7 Disolución acuosa saturada de cloruro de sodio.
57.3.8 Tierra Tonsil AC o similar.
57.3.9 Disolución de fenolftaleína en metanol al 1 por 100.
57.3.10 Iodo resublimado.
57.3.11 Disolución acuosa de hidróxido de sodio 2N.
57.3.12 Sulfato de sodio anhidro.
57.4 Procedimiento.
57.4.1 Preparación de la muestra.
En el caso de tratarse de aceites de oliva vírgenes de calidad comestible, aceites de oliva refinados o aceites de orujo refinados, y sus mezclas, no será necesaria ninguna preparación previa, limitándose a efectuar una filtración por papel de filtro en el caso de que la muestra estuviese turbia o contuviera humedad. La muestra para el ensayo debe estar limpia y transparente.
Si se trata de aceites de oliva vírgenes no comestibles o aceites de orujo brutos deberán someterse las muestras a un proceso previo de purificación, consistente en una neutralización y una decoloración.
Neutralización. En una ampolla de extracción de 500 ml introducir unos 20 g de muestra, 100 ml de hexano, 50 ml de etanol, unas gotas de disolución alcohólica de fenolftaleína al 1 por 100, y el volumen calculado de disolución de hidróxido de sodio 2N equivalente a la acidez libre de la muestra, medido con una bureta graduada en décimas de ml.
Si la fenolftaleína no acusase reacción alcalina, deberá adicionarse más disolución de hidróxido de sodio, gota a gota, agitando después de cada adición, hasta conseguir el viraje del indicador.
Agitar la mezcla enérgicamente, agregar 50 ml de agua destilada, agitar nuevamente y dejar en reposo hasta que la mezcla se separe en dos capas bien definidas y extraer por la llave de capa hidroalcohólica conteniendo los jabones.
La disolución de hexano conteniendo la grasa neutra se lava con porciones sucesivas de 25 ml de una mezcla de etanol de 96 y agua destilada 1:1 (v/v), hasta que el líquido de lavado no se coloree en rosa con la fenolftaleína.
La disolución de hexano, libre de jabones, se deseca agregando 3 g-4 g de sulfato de sodio anhidro; filtrar sobre un matraz de 250 ml, redondo, de fondo plano, y evaporar el disolvente en un evaporador rotatorio al vacío.
Decoloración. Tomar 10 ml de aceite neutralizado, que se introduce en una probeta de 25 ml, con tapón esmerilado. Seguidamente agregar 1 g de tierra decolorante y agitar fuertemente durante unos treinta segundos, filtrándose, a continuación, por un filtro de pliegues.
57.4.2 Separación de los triglicéridos más insaturados.
Los ensayos se deben hacer por duplicado, simultaneándolos con un aceite testigo cuyos resultados nos sean previamente conocidos (57.6.2).
De la muestra preparada, como se indica en el 57.4.1, en sendos tubos de ensayo (57.2.5), pesar 0,5 g ± 0,01 g, añadir 20 ml de la mezcla metanol-acetona 7/3 (v/v), recién preparada, taponar y agitar fuertemente. Seguidamente, introducirlos en el arcón de congelación, colocándolos en un vaso u otro dispositivo que pueda servir de soporte, situándolos en la plataforma citada en 52.2.2; la temperatura del arcón habrá sido previamente regulada a -22 ºC ± 1 ºC. Cerrar la tapa del arcón, debiendo permanecer así quince horas.
Aproximadamente una hora antes de que transcurra el período indicado de quince horas, colocar en la plataforma del arcón, al lado de los recipientes de cristalización, dos matraces de 50 ml, de forma de corazón, y dos embudos de vidrio con sus papeles de filtro, protegidos ambos por un vidrio de reloj.
Pasadas las quince horas, levantar la tapa del arcón, procediéndose a separar por filtración los triglicéridos cristalizados de la solución conteniendo los triglicéridos más insaturados, realizándose esta operación en la misma plataforma donde se encuentran los recipientes.
Colocar el embudo con filtro en la boca del matraz, y sin agitar el recipiente conteniendo el problema decantar el líquido sobre el filtro, evitando, en lo posible, el arrastre del sólido cristalizado, y procurando una filtración rápida, no siendo necesario agotar el contenido del tubo de ensayo. En esta operación los tubos de ensayo se deben coger por el cuello esmerilado, cuidándose no sacarlos del recinto del arcón.
Una vez se da por terminada la filtración se retiran los embudos de los matraces antes de sacarlos del arcón, se recogen los matraces y se evapora el disolvente en evaporador rotatorio al vacío.
57.4.3 Purificación del extracto. Disolver el residuo en 0,25 ml de hexano y depositarlo en forma de banda en una placa de CCF con gel de sílice, de 20 x 20 cm y un espesor de capa de 0,25 mm previamente activada por calentamiento en estufa a 105 ºC durante unos quince minutos.
El desarrollo se realiza en cubeta cromatográfica, con una mezcla hexano-éter etílico 8:2 (v/v), recientemente preparada, hasta que el frente del disolvente quede a unos 4 cm del extremo superior de la placa.
Concluido el desarrollo, tomar una placa cromatográfica limpia, sin recubrimiento de sílice, y se la coloca encima de la placa desarrollada, dejando al descubierto una banda lateral de unos 3 cm de ancho, fijando el conjunto con una pinza.
Se introduce el conjunto de las dos placas en una cubeta saturada de vapores de iodo, debiendo aparecer una banda ancha correspondiente a los triglicéridos, claramente separada de otros componentes minoritarios que pueden interferir.
57.4.4 Metilación de los ácidos grasos. Raspar la banda de los triglicéridos recogiéndose el raspado en un papel satinado, pasando el polvo al matraz de metilación (57.2.13).
Agregar al matraz 10 ml de disolución de metilato de sodio y hervir en baño María a reflujo, durante cinco minutos.
Retirar el matraz del baño, agregar una gota de solución de fenolftaleína y a continuación disolución metanólica de clorhídrico, hasta reacción ácida y un volumen igual al consumido para neutralizar, como exceso, hirviendo de nuevo diez minutos.
Enfriar la disolución con los ésteres metílicos, agregados seguidamente al matraz 0,25 ml de hexano y 15 ml de solución acuosa saturada de cloruro de sodio. Agitar fuertemente, adicionando a continuación, la cantidad necesaria de la misma solución salina, para que la capa de hexano se sitúe en el cuello del matraz.
Dejar reposar unos minutos para que la capa de hexano se separe limpia en el cuello del matraz, pasándose seguidamente a efectuar el análisis de los ácidos grasos por cromatografía gaseosa.
Se realiza también la metilación de la muestra de aceites originales utilizado para el fraccionamiento, siguiendo el método oficial número 41.
57.4.5 Ácidos grasos por cromatografía gaseosa. Según método oficial número 41.
57.5. Expresión de los resultados.
Sea x el contenido en ácido linoleico de la muestra original encontrado por análisis gas-cromatográfico; e y el contenido en ácido linoleico de la fracción más insaturada.
En la tabla I se entiende para cada valor de x el valor previsible para y, en el supuesto de que se trate de un aceite genuino, teniendo en cuenta la dispersión real de resultados, calculado con un nivel de probabilidad de 0,99.
Para valores de x intermedios, no incluidos en la tabla, se calculará el valor previsible de y por interpolación entre los dos valores más próximos.
Si el valor encontrado para el ácido linoleico en la fracción más insaturada es superior a la cifra deducida de la tabla se puede considerar a la muestra adulterada; para valores inferiores o iguales hay que considerarla como genuina (57.6.3 y 57.6.4).
Tabla I
Valores máximos previsibles en aceites de oliva genuinos para el contenido de ácido linoleico de la fracción más insaturada, en función del contenido en ácido linoleico del aceite original.
| X | Y |
|---|---|
| 2,000 | 32,633 |
| 4,000 | 35,589 |
| 6,000 | 38,589 |
| 8,000 | 41,640 |
| 10,000 | 44,747 |
| 12,000 | 47,907 |
| 14,000 | 51,120 |
| 16,000 | 54,380 |
| 18,000 | 57.686 |
| 20,000 | 61,029 |
57.6 Observaciones.
57.6.1 Es esencial, para una aplicación correcta del procedimiento, la realización de las operaciones de cristalización y de separación de la parte cristalizada a una determinada temperatura y en condiciones que sean reproductibles entre laboratorios.
57.6.2 La utilización de la muestra testigo, de respuesta previamente conocida, en el ensayo no es indispensable, teniendo solamente por objeto el garantizar no haberse producido ningún Fallo en el curso de la cristalización por causas que podrían pasar desapercibidas al operador, falseando los resultados.
57.6.3 Para valores intermedios de x no incluidos en la tabla a función y puede considerarse lineal dentro de cada intervalo, efectuándose el cálculo por interpolación.
No obstante, el analista que desee operar con mayor seguridad puede calcular la ecuación de la línea de regresión, a partir de datos experimentales propios, obtenidos con muestras patrones de garantía; como, asimismo, los límites de confianza asignables a estos parámetros.
57.6.4 En la evaluación de resultados hay que tener en cuenta que la sensibilidad del procedimiento, en orden al límite de detección de aceite semi-secante en aceite de oliva, depende, fundamentalmente, del contenido de ambos aceites en ácidos polisaturados. En los análisis colaborativos realizados con mezclas, en distintas proporciones, de un aceite de girasol y un aceite de oliva de características normales se puso de manifiesto un límite de identificación situado entre el 2-3 por 100 de girasol. Pero en circunstancias más favorables el límite de detección puede descender por bajo del 1 por 100, pudiéndose tratar de una contaminación.
57.6.5 Se puede utilizar un sistema de congelación distinto al descrito siempre que asegure las mismas condiciones de trabajo.
57.7 Referencias bibliográficas.
‒ Instituto Español de Normalización. UNE 55-132-83.
58. Determinación del eritrodiol
58.1 Principio.
Determinados aceites vegetales, corno el aceite de oliva, orujo de aceituna, pepita de uva, etc., contienen en su insaponificable dialcoholes terpénicos. El componente mayoritario de éstos es el eritrodiol.
Su determinación consta de la extracción del insaponificable y su posterior fraccionamiento en cromatografía en capa fina, recuperándose juntamente las fracciones de esteroles y dialcoholes terpénicos, que se silanizan, analizándose seguidamente por cromatografía en fase gaseosa.
58.2 Material y aparatos
58.2.1 El material descrito en la norma UNE 55.004 para la separación del insaponificable por el método del éter etílico.
58.2.2 Columna cromatográfica de unos 30 cm de longitud y 15 mm de diámetro, preferiblemente con placa filtrante de vidrio, porosidad número 2.
58.2.3 Equipo para la preparación de placas.
58.2.4 Placas de vidrio de 20 por 20 cm.
58.2.5 Cubeta de vidrio con su tapa para el desarrollo de placas 20 por 20 cm.
58.2.6 Soporte metálico para la conservación y transporte de las placas.
58.2.7 Cámara para la conservación de las placas.
58.2.8 Estufa para la desecación de las placas.
58.2.9 Evaporador rotatorio con vatio y baño de agua regulable a 35 ºC.
58.2.10 Micropipeta o microjeringa capaz de liberación gotitas de 0,3-0,4 µl (58.5.2).
58.2.11 Pulverizador para la aplicación del reactivo de revelado.
58.2.12 Tubo cromatográfico representado en la figura, destinado a la elución de las fracciones separadas en la cromatografía en capa fina.
58.2.13 Lámpara ultravioleta para el examen de las placas
58.2.14 Equipo de cromatógrafo gaseosa, constituido fundamentalmente por el cromatógrafo y el registrador, provisto o no de integrador, con todos los elementos auxiliares para su manejo, tales como tubos de gases a presión, medidor de flujo, etc.
El cromatógrafo irá equipado con un sistema de detección de ionización, de llama, provisto de inyector y columna de vidrio.
58.2.15 Columna de vidrio de unos 2 m de longitud, con diámetro exterior de 7,5 mm y diámetro interior de 2,5 mm, con relleno de una fenil-metil silicona al 50 por 100 (OV-17), al 5 por 100 sobre Supelconport 80/100 mallas o Chromosorb W-HP de la misma granulometría.
58.2.16 Microjeringa de 5-10 µl, con graduación en 0,1 µl o 0,2 mI.
58.2.17 Matracitos en forma de corazón de 5 y 15 de capacidad y tapón esmerilado.
58.3 Reactivos
58.3.1 Los reactivos mencionados en la norma UNE 55.004, necesarios para la separación del insaponificable por el método del éter etílico.
58.3.2 Resina intercambiadora de iones grado analítico, Amberlite MB-3 u otra de características similares (58.5.5).
58.3.3 Gel de sílice G Merck u otro producto de características similares.
58.3.4 Solución de 2' -7' diclorufluoresceina al 0,1 por 100 en etanol del 90 por 100. Se puede emplear cualquier otro colorante que no tenga reacción con los esteroles, por ejemplo la Rodamina 6G.
58.3.5 Éter etílico de calidad «reactivo análisis».
58.3.6 Hexano de calidad «para cromatografía».
58.3.7 Solvente de desarrollo hexano-éter etílico 2:1 (58.5.6).
58.3.8 Éter isopropílico, calidad «reactivo análisis».
58.3.9 Colesterol purísimo al 1 por 100 en éter isopropílico.
58.3.10 Algodón de vidrio calidad cromatográfica.
58.3.11 Solución al 1 por 100 (m/v), en éter isopropílico de β-sitosterol, patrón cromatográfico, con una pureza mínima del
98 por 100.
58.3.12 Reactivo de silanización. En un tubo de ensayo Sovirel, con tapón de rosca, previamente desecado, se introducen 9 ml de piridina recientemente destilada sobre hidróxido sódico y desecada con criba molecular de 4 A. Se adicionan 3 ml de hexametildisilazano y 1 ml de trimetil clorosilano, ambos de calidad purísimos.
58.3.13 Solución de éter isopropílico de betulina, con una riqueza mínima del 99 por 100, conteniendo 50 mg/100 ml.
58.4 Procedimiento
58.4.1 Aislamiento de los esteroles y los dialcoholes terpénicos.
Aislamiento del insaponificable. Extraer el insaponificable de 5 g de muestra, siguiendo el método descrito en UNE 55.004 (método del óxido dietílico), hasta la adición de acetona, eliminación del disolvente volátil en baño de agua caliente (58.5.7 y 58.5.8).
En el caso de que se quiera efectuar la cuantificación empleando la betulina como patrón interno, una vez pesada la muestra de grasa, con una precisión del centigramo, se adiciona al matraz 5 ml exactamente medidos de la solución patrón de betulina (58.3.13) conteniendo 0,5 mg/ml, procediendo, a continuación, de la forma indicada en el párrafo anterior.
Purificación del insaponificable. Una vez eliminado el disolvente y los últimos restos de aguas por arrastre con acetona, se disuelve la materia insaponificable en el mismo matraz en 20 ml de óxido dietílico. Se agregan 6 g de resina intercambiadora de iones previamente desecada e impregnada de óxido dietílico como se indica en el punto anterior, y se mantiene en agitación en un agitador magnético durante 20-30 minutos. La solución atérea se filtra por papel, recogiendo el filtrado en un matracito de 25 ml en dos porciones, eliminando cada vez el disolvente en evaporador rotatorio. Una vez filtrada toda la disolución, se lavan el matraz y el filtro con tres porciones de 3-4 ml cada una del mismo disolvente, eliminando todo por evaporación y dejando el insaponificable seco.
58.4.2 Fraccionamiento del insaponificable.
Preparación de las cromatoplacas. Colocar las placas en el extendedor ocupando toda la longitud disponible, empleándose generalmente 4 placas de 20 x 20 cm y 2 placas de 20 x 5 cm.
Limpiar cuidadosamente la superficie de las placas con hexano, etanol acetona, con el fin de asegurar la eliminación completa de todo residuo de grasa.
En un matraz Erlenmeyer de 250 ml se introduce 30 g de gel de sílice con aglutinante y 60 ml de agua destilada. Se tapa el matraz y se agita fuertemente durante un minuto, con el fin de asegurar la homogeneidad más perfecta posible de la pasta. Inmediatamente, se pasa la pasta al aplicador y se extiende sobre las placas, con un espesor de 0,25 mm. El tiempo empleado en la preparación de la pasta y su extendido sobre las placas no deberá exceder de 4 minutos.
Se dejan las placas en reposo en el extendedor para su secado al aire, hasta observar la superficie seca. A continuación, se retiran del extendedor y se colocan en el cortaplacas, introduciéndolo en una estufa de desecación regulada a 105 ºC-110 ºC, donde se mantiene una hora.
Terminada la activación, se introducen las placas en la cámara (58.2.7), para su conservación.
Preparación de la cubeta de desarrollo Introducir en la cubeta de desarrollo el volumen necesario del líquido de desarrollo (58.3.7 y 58.5.6), para alcanzar una altura de unos 10 mm. Se tapa la cubeta y se espera el tiempo necesario para conseguir el equilibrio entre las dos fases líquido/vapor, siendo suficiente, por lo general, una hora.
Cromatografía del insaponificable. El insaponificable purificado y seco, contenido en el matraz de 25 ml, se disuelve en 5 ml de éter isopropílico. De 2 a 2,5 ml de esta disolución se introducen en un matracito de 5 ml (58.2.17), eliminando el disolvente con una corriente de nitrógeno. Disolver el residuo en 200 a 500 µl de éter isopropílico.
En una placa cromatográfica, preparada y activa como se ha indicado en el apartado de preparación de las cromatoplacas, se deposita en gotitas finas, valiéndose de una micropipeta o microjeringa, la totalidad de la disolución anterior del insaponificable, cubriendo una línea situada a 2 cm del borde inferior de la placa y a 2,5 cm de los extremos derecho e izquierdo. La distribución de la disolución deberá hacerse de la manera más uniforme posible, efectuando las pasadas que sean necesarias para depositar la totalidad, pero esperando a cada nueva aplicación que haya vaporizado el disolvente.
A 1 cm del extremo derecho de la línea de aplicación del problema y a 1 cm del extremo izquierdo, se depositan 15-20 µl de la solución de colesterol (58.3.9).
Introducir inmediatamente la placa en la cubeta de desarrollo, preparada como se indica anteriormente. Tapar la cubeta y dejar que efectúe el desarrollo hasta que el frente del disolvente se sitúe a unos 2 cm del borde superior de la placa. Sacar la placa de la cubeta y dejarla secar al aire, para lo que son suficientes unos 2 o 3 minutos. Se pulveriza la placa con el reactivo de revelado (58.3.4) y se examina a la luz ultravioleta (356 nm).
Tomando como referencia las dos manchas de colesterol, se fija la posición de la banda de los esteroles y la de los dialcoholes triterpénicos que se sitúan por debajo de los esteroles, en una posición perfectamente diferenciada (58.5.10).
La banda horizontal de sílice de la placa delimitada por las dos marcas comprendiendo los esteroles y los dialcoholes triterpénicos, se rasca con una espátula recogiendo el polvo en un papel satinado y pasándolo seguidamente a la columnita de elución (58.2.12), que se prepara de la forma siguiente:
Se coloca un tapón de algodón de vidrio en el extremo del cuerpo intermedio de la columna, con un diámetro interno de 6 mm, ocupando una altura de 30 mm. Verter encima de este tapón la sílice raspada de la placa, y completar con gel de sílice sin utilizar hasta llenar completamente el tubo de 6 mm de diámetro.
Sujetar el tubo en posición vertical en un soporte, poniendo debajo como colector un matracito de 15 ml de capacidad (58.2.17). Se vierte por la boca de la columna éter isopropílico, en porciones de 1 ml, esperando para efectuar cada adición que haya penetrado en la columna de sílice la porción anterior. Se efectuarán 6 adiciones con un total de 6 ml. Terminada esta operación, se evapora el disolvente en evaporador rotatorio hasta sequedad.
58.4.3 Cromatografía gaseosa.
Silanización del problema. Al matracito conteniendo la fracción de esteroles y los dialcoholes triterpénicos, se adicionan 200 µI del reactivo de silanización (58.3.12), cuidando que la jeringa y todos los recipientes utilizados estén perfectamente secos. Para conseguir esto, resulta conveniente, inmediatamente antes del uso, enjugarlos con acetona anhidra, que se escurre, evaporando el resto con una corriente de aire seco.
Se tapa el matracito, agitándose suavemente mediante un movimiento de rotación durante 1 o 2 minutos. Se deja en reposo unos 20 minutos, quedando la solución lista para su inyección en el cromatógrafo.
Condiciones de trabajo. No se puede indicar condiciones estrictas de trabajo, dependiendo fundamentalmente del comportamiento de la columna, características del apartado, etcétera. Puede considerarse que la operación marcha satisfactoriamente si se registra para β-sitosterol un pico simétrico, sin la aparición de «colas» y la resolución del para-campesterol-estigmasterol es, como mínimo, de 0,8 (58.5.11 y 58.5.12). Si estas condiciones mínimas no se cumpliesen incluso modificando las condiciones de trabajo dentro de límites aceptables, sería necesario rechazar la columna sustituyéndola por otra.
A título de orientación, se indican a continuación las condiciones de trabajo alrededor de las cuales se suelen encontrar los valores óptimos, debiendo ser modificados, en cada caso, hasta encontrar una respuesta satisfactoria.
‒ Temperatura de la columna: 240 ºC-250 ºC.
‒ Temperatura del inyector: 290 ºC.
‒ Flujo del gas portador (N2): 30-50 ml/minuto.
Se inyecta, aproximadamente, 0,5-0,6µ 1 de la solución silanizada, utilizando la sensibilidad necesaria para que el pico del β-sitosterol llegue casi al extremo del papel registrador, efectuándose preferiblemente la totalidad del registro del cromatograma a la misma sensibilidad. En todo caso, no deberá existir más de una atenuación ente los registros del β-sitosterol y el del eritrodiol.
Identificación de los picos. EI pico correspondiente al β-sitosterol se identifica muy fácilmente por un operador con experiencia, por ser el componente mayoritario de la fracción. En caso de duda, podría efectuarse la identificación inyectando 0,5-0,6 µl de la solución patrón de β-sitosterol (58.3.11), previamente silanizado como se indica en (58.4.3). El eritrodiol se registra después del β-sitosterol, con un tiempo de retención en relación a éste de 1,3 a 1,5 operando en las condiciones descritas.
Se registran, también, otros dialcoholes triterpénicos, pero en una cuantía muy inferior a la del eritrodiol.
La betulina se registra en relación al β-sitosterol con un tiempo de retención aproximadamente de 1,6.
Determinaciones cuantitativas. (58.5.2).
Cuantificación relativa al β-sitosterol. Se calcula la respuesta del detector en los picos del β-sitosterol y del eritrodiol, multiplicando en cada uno de ellos su altura por el ancho a la mitad de la altura, v en el caso de haberse efectuado los registros a sensibilidades diferentes, se referirán ambos a una misma sensibilidad (58.5.9).

AE = área correspondiente al eritrodiol.
AS = área correspondiente al β-sitosterol.
Cuantificación con patrón interno Se calculan las respuestas del detector para los picos del eritrodiol y de la betulina, siguiendo el mismo criterio que se indica anteriormente.

AE = Área correspondiente al eritrodiol.
Ap = Área correspondiente a la betulina.
mp = Peso del patrón interno utilizado, expresado en mg.
ms = Peso tomado de la muestra de aceite, en g.
KEp = Factor de respuesta del eritrodiol frente al patrón utilizado.
Utilizándose la betulina como patrón interno, y a las concentraciones que se indican en la metódica, el factor de respuesta es, prácticamente, igual a la unidad.
58.5 Observaciones.
58.5.1 La purificación del insaponificable mediante trata con resina no es indispensable para la aplicación de la metódica, teniendo únicamente por finalidad la eliminación de los últimos restos de jabones y/o ácidos grasos que pudieran interferir en el fraccionamiento por capa fina. Por tanto, este tratamiento puede suprimirse sin grave inconveniente y siempre que no surjan dificultades en el fraccionamiento del insaponificable derivados de la presencia de los productos que la resina debe eliminar.
58.5.2 En los casos de aceites de oliva y de orujo de aceituna, en los que el contenido de eritrodiol en relación al β-sitosterol vaya a utilizarse como criterio de diferenciación, debe tomarse el denominador β-sitosterol «aparente», dado por la suma del β-sitosterol y Δ5-avenasterol. Para este fin, es recomendable la utilización como fase estacionaria del SE-30, va que, con esta fase, estos dos esteroles se registran en un solo pico.
58.5.3 La utilización de este tubo no es imprescindible, pudiendo ser sustituido por una varilla hueca de 7-8 mm de diámetro interior, y una longitud aproximada de 250 mm, con una estrangulación en el extremo para un tapón de lana de vidrio de 20-30 mm.
58.5.4 Tanto la fase fija como los soportes que se señalan, se hace sólo a título orientativo por haberse experimentado con resultados satisfactorios. Sin embargo, pueden ser sustituidos por otros rellenos que podrán dar resultados análogos, o incluso, mejores, siempre que la columna satisfaga los criterios de eficacia que se dan en el apartado (58.5.3).
Con columnas de OV-17 o SE-30 puede producirse el solapamiento del eritrodiol con el Δ5-avenasterol que, en determinados casos, puede estar presente en cantidades significativas. Estos esteroles pueden separarse perfectamente con columna capilar de OV-17 (58.5.2).
58.5.5 Antes de su utilización para la purificación del insaponificable, la resina deberá ser sometida al tratamiento siguiente:
6-7 g de resina se introduce en una columna cromatográfica (58.2.1), agregándose 100 ml de metanol anhidro que se hacen pasar a una velocidad aproximada de una gota por segundo. Una vez pasada la totalidad del metano, se hace pasar por la columna una corriente de aire con el fin de eliminar los restos de metanol. La resina así desecada puede conservarse en un frasco herméticamente cerrado.
La resina se hidrata con mucha facilidad, reteniendo grandes cantidades de agua. Por lo tanto, en algunos casos, la cantidad indicada de metanol puede ser insuficiente para lograr la deshidratación total de la resina, siendo necesario un lavado más prolongado, utilizando mayor cantidad de metanol.
Inmediatamente antes de su utilización, la cantidad necesaria de resina (6 g), se pone en suspensión en 10-15 ml de óxido dietílico, manteniéndola así unos quince minutos, agitando suavemente de vez en cuando.
58.5.6 En lugar de la mezcla hexano-óxido dietílico para el desarrollo de la placa, pueden utilizarse otros disolventes con resultados análogos, y siempre que se consiga una separación clara de los esteroles y los dialcoholes tritarpénicos que permita su localización en la placa, para su raspado y recuperación cuantitativa. Se han recomendado por algunos experimentadores el cloroformo; benceno-acetona (95:5); heptano normal-acetona (85:15); etcétera.
58.5.7 En el caso de que la solución etérea del insaponificable contuviese mucha agua, lo cual es lo más frecuente, es conveniente proceder a una desecación con sulfato sódico. Para ello, se adiciona a la solución contenida en la ampolla de extracción 8-10 g de sulfato sódico anhidro, agitándose y dejando en reposo durante treinta-cuarenta y cinco minutos. Seguidamente, se filtra la solución, que se recoge en el matraz en el que se vaya a efectuar la destilación. La ampolla y el filtro se lavan con tres porciones de óxido dietílico de 5 ml cada una.
58.5.8 Todas estas operaciones de evaporación se pueden realizar con mayor rapidez y comodidad utilizando un evaporador rotatorio de vacío. En cualquier caso, la disolución del insaponificable no deberá ser calentada por encima de los 40 ºC.
58.5.9 En las operaciones de cuantificación de los picos, puede emplearse un integrador electrónico, sustituyendo en este caso los valores de áreas por el número de «cuentas» dado por el integrador.
58.5.10 En el caso de que se disponga de eritrodiol de la pureza suficiente para su utilización como patrón cualitativo, se adicionará a la solución testigo de colesterol el 1 por 100 de eritrodiol, sirviendo de orientación la mancha obtenida en el cromatograma para fijar la posición del eritrodiol en el problema. Sin embargo, esto no es indispensable, pues la posición del eritrodiol es fácil de fijar a partir de la de los esteroles.
58.5.11 La resolución viene dada por la fórmula:

d2 y d1 = distancias en el registro del máximo de los picos al frente de salida del disolvente.
b2 y b1 = bases de los picos.
58.5.12 Para verificar la condición impuesta relativa a la resolución del campesterol y estigmasterol, no es necesario disponer de patrones de estos productos, siendo muy fácil su identificación en el cromatograma obtenido con la fracción de esteroles. En las condiciones operativas indicadas, el campesterol tiene un tiempo de retención, referido al β-sitosterol de 0,80 aproximadamente y el estigmasterol de 0,82.
Columna de elución
Cotas en milímetros
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




