Agencia Estatal Boletín Oficial del Estado






Excelentísimos señores: De acuerdo con lo dispuesto en el Decreto 1043/1973, de 17 de mayo, por el que se regula la normalización de productos ganaderos en el mercado interior, como medio de corregir determinados defectos de los sistemas de comercialización, así como de garantizar al consumidor la calidad de los productos que adquiere y orientar la producción por cauces cualitativos, vistos los acuerdos del F. O. R. P. P. A., y a propuesta de los Ministros de la Gobernación, de Agricultura y de Comercio, esta Presidencia del Gobierno dispone:
Se aprueba la norma para mantequilla que a continuación se transcribe:
1. NOMBRE DE LOS PRODUCTOS
Mantequilla y mantequilla de suero.
2. DESCRIPCION O DEFINICION
Mantequilla es el producto graso obtenido exclusivamente de leche o nata de vaca higienizadas.
Mantequilla de suero es el producto graso obtenido del suero higienizado que no contenga ninguna otra grasa más que la de leche de vaca.
3. AMBITO DE APLICACION,
La presente norma se aplica a la mantequilla y mantequilla de suero que se fabriquen y/o comercialicen dentro del territorio nacional, quedando exceptuada la «Mantequilla de Soria» y/o «Mantequilla azucarada».
Lo dispuesto en la presente norma no exime del cumplimiento de lo estipulado en otras disposiciones de distinto carácter.
4. FACTORES ESENCIALES DE COMPOSICION Y CALIDAD
4.1 Organolépticos.
La mantequilla dispuesta para su venta deberá presentar las siguientes características:
4.1.1 Consistencia sólida y homogénea.
4.1.2 Color amarillo uniforme más o menos intenso.
4.1.3 Sabor y olor característicos.
4.2 Intrínsecos (materias primas).
4.2.1 Contenido, mínimo de materia grasa, de la leche de procedencia, 80 por 100 m/m. (masa/masa).
4.2.1.1 Las características físico-químicas de la grasa, estarán comprendidas entre los siguientes valores:
Indice de refracción a + 40° C, de 1,4557 a 1,4540.
Indice de Reichert, de 32 a 25.
Indice de Poienske, de 4 a 1.
Indice.de Kirchner, de 27 a 19.
4.2.2 Contenido máximo de extracto seco magro, de la leche de procedencia, 2 por 100 m/m. (masa/masa).
4.2.3 Contenido máximo de agua, 16 por 100 m/m. (masa/ masa).
4.3 Añadidos.
4.3.1 Cloruro sódico, en dosis máximas del 5 por 100 m/m. (masa/masa).
4.3.2 Fermentos lácticos, en dosis limitadas por la correcta práctica de fabricación.
4.4 Sanitarios.
4.4.1 Ausencia total de gérmenes patógenos.
4.4.2 Ausencia de gérmenes coliformes en 0,1 gramos.
4.4.3 Ausencia de Escherichia Coli en 0,1 gr.
4.4.4 Recuento de mohos, máximo. 10 por gr.
4.4.5 Recuento de levaduras, máximo 100 por gr.
4.4.6 Microorganismos lipolíticos, máximo 10 por gr.
4.4.7 Prueba de la fosfatasa negativa.
4.4.8 Ausencia de óxidos alcalino-térreos en la fracción magra.
5. ADITIVOS ALIMENTARIOS
5.1 Colorantes.
5.1.1 Bixina o bija o achiote. Cantidad limitada por la correcta practica de fabricación.
5.1.2 Beta-caroteno (forma trans.). Cantidad limitada por la correcta práctica de fabricación.
5.1.3 Curcumina (amarillo N-1, color Índex 75.300). Cantidad limitada por la correcta práctica de fabricación.
5.1.4 Aquellos otros que autorice la Dirección General de Sanidad.
5.2 Sales neutralizantes.
La adición de las siguientes sales, para el ajuste del pH, viene limitada por separado o en su conjunto al 0,2 por 100 m/m. (masa/masa), expresado dicho porcentaje en masa de las sustancias anhidras, referido a masa del producto total.
5.2.1 Ortofosfato sódico.
5.2.2 Carbonato sódico.
5.2.3 Bicarbonato sódico.
5.2.4 Hidróxido sódico.
5.2.5 Hidróxido cálcico.
5.2.6 Aquellas otras que autorice la Dirección General de Sanidad.
5.3 Agentes conservadores, antioxidantes y sinérgicos. Podrán emplearse los autorizados por la Dirección General de Sanidad para el uso específico en mantequillas, previa solicitud con este fin.
6. PROHIBICIONES
En el curso de la manipulación, tratamiento y venta de la mantequilla, se prohíbe:
6.1 La mezcla con margarina y/o cualquier otra clase de grasa.
6.2 La adición o presencia de cualquier materia extraña no autorizada, en la presente norma.
6.3 La adición o presencia de colorantes y neutralizantes no incluido en los apartados 5.1 y 5.2, así como la de otros aditivos no autorizados para este producto.
6.4 La adición de fermentos distintos a los lácticos.
6.5 Almacenar, elaborar o manipular en las fábricas o almacenes de mantequilla, otras grasas alimenticias o industriales, así como productos que puedan ser destinados a su adulteración.
6.6 La venta al público a granel y la venta fraccionada de bloques o paquetes de mantequilla en los establecimientos de venta al público, así como su servicio para consumo en los establecimientos del ramo de hostelería.
6.7 La tenencia de mantequilla a granel o la de bloque o paquetes fraccionados y envases abiertos en los locales de venta al público, exceptuándose la de uso propio o de cocina, en establecimientos de la industria alimentaria y en el ramo de hostelería.
6.8 Todo empleo de indicaciones o presentación de etiquetas, envases, embalajes, documentos comerciales y medios de publicidad que sean susceptibles de crear en el ánimo del consumidor cualquier clase de confusión sobre la naturaleza, composición, peso neto u origen del producto.
6.9 La venta de mantequilla con sabores y olores extraños.
6.10 La venta de mantequilla con manchas de mohos y/o levaduras o de cualquier otra procedencia.
6.11 La, impresión o litografía en la cara interna del envase o envoltura, que esté en contacto con el alimento.
7. ENVASADO
7.1 La mantequilla solamente podrá envasarse en las industrias legalmente autorizadas para este fin.
7.2 La mantequilla se presentará al consumidor en bloques o formatos de cualquier medida o peso obligatoriamente envuelta en papel sulfurizado o metalizado o envasada en recipientes de hojalata, vidrio o plástico autorizados. Asimismo podrá utilizarse cualquier otro material, previamente autorizado por la Dirección General de Sanidad.
7.3 En el peso neto individual se admitirá una tolerancia del 5 por 100 sobre el peso en origen para los envases iguales o inferiores a cien gramos y del 3 por 100 para los superiores a este peso.
8. ETIQUETADO
El etiquetado de la mantequilla llevará en los envases originales, así como en el embalaje, en idioma español y en caracteres bien visibles, las siguientes especificaciones.
8.1 Denominación del producto, que será mantequilla o mantequilla de suero, según corresponda.
8.2 La expresión «salada» o «con sal», en el caso de que se haya añadido cloruro sódico.
8.3 El peso neto en origen expresado en unidades del sistema métrico decimal.
8.4 Nombre y dirección del fabricante o envasador o del importador, distribuidor o vendedor del alimento en su caso. El nombre y dirección del fabricante podrá ser sustituido por su marca comercial registrada.
8.5 Fecha o su clave del envasado definitivo, bien impreso o perforado, y número de registro del envasador en la Dirección General de Sanidad para las de fabricación nacional.
La toma de muestras y determinaciones analíticas se realizarán de acuerdo con lo dispuesto en el Anejo Unico de esta Orden.
La presente Orden entrará en vigor en todo el territorio nacional a los seis meses de su publicación en el «Boletín Oficial del Estado».
De acuerdo con lo dispuesto en el artículo undécimo del Decreto 1043/1973, de 17 de mayo, los Ministerios de la Gobernación, de Agricultura y de Comercio, ejercerán las funciones de control y vigilancia de lo dispuesto en la presente Orden, dentro del ámbito de sus respectivas competencias y a través de los órganos administrativos correspondientes que se coordinarán en sus actuaciones.
Lo digo a VV. EE. a los procedentes efectos.
Dios guarde a VV. EE.
Madrid, 7 de enero de 1975.
CARRO
Excmos. Sres. Ministros de la Gobernación, de Agricultura y de Comercio.
1. TOMA DE MUESTRA
Obtención a partir de una unidad (recipiente o paquete) de una parte (submuestra) que sea lo más representativa posible de dicha unidad.
1.1 Materiales.
Podrán utilizarse los siguientes materiales:
– Sondas de acero inoxidable con longitud suficiente para alcanzar diagonalmente la base del recipiente y con un diámetro igual o superior a 30 milímetros.
– Espátulas o cuchillos, de acero inoxidable para retinar parte de la muestra tomada con la sonda.
– Recipientes cilíndricos de boca ancha, de vidrio o de acero inoxidable, esterilizables, de capacidad adecuada al tamaño de la muestra a tomar y que cerrarán herméticamente.
Todos los materiales utilizados deberán estar secos y limpios y no deberán comunicar olores ni sabores extraños. Si la muestra se destina a análisis microbiológicos, el material se esterilizará por tratamiento con alcohol, seguido de flameado, o por inmersión en agua a + 100° C, por lo menos, treinta segundos, y se enfriarán a temperatura ambiente antes de su uso.
1.2 Técnica.
Se tomará un número suficiente de muestras parciales para que el peso de la muestra sea, por lo menos, de 200 gramos. El número de muestras tomadas será el que determina la legislación vigente.
Según la forma y peso de la mantequilla, se aplicará una de las técnicas siguientes:
a) Mantequilla en bloques, o recipientes autorizados de peso unitario superior a un kilogramo.
b) Mantequilla en recipientes autorizados de peso unitario comprendido entre 0,25 kilogramos, y un kilogramo.
c) Mantequilla en recipientes autorizados de peso unitario inferior a 0,25 kilogramos.
a) Mantequilla en bloques, o recipientes autorizados de peso unitario superior a un kilogramo:
Se harán dos sondajes de la mantequilla. Uno de estos se obtendrá introduciendo una sonda en diagonal a través del bloque de mantequilla desde una esquina de la extremidad abierta del recipiente. El segundo sondaje se obtendrá introduciendo la sonda verticalmente desde un punto arbitrario de la superficie hasta la base del recipiente La muestra comprenderá porciones tomadas de diferentes puntos de los dos sondajes para dar un peso total mínimo de 200 gramos. Los recipientes para las muestras no se llenarán menos de dos tercios ni más de nueve décimos de su-capacidad. Inmediatamente después de cerrados, los recipientes- que contienen la mantequilla serán envueltos en papel, y almacenados en sitio oscuro. La mantequilla no estará en contacto ni con el papel ni con ninguna superficie absorbente de agua o grasa.
b) Mantequilla en recipientes autorizados de peso unitario comprendido entre 0,25 kilogramos y un kilogramo:
Dividir en cuatro partes aproximadamente iguales las unidades, y elegir como muestra dos partes opuestas o la fracción correspondiente de dos partes opuestas para dar un peso total mínimo de 200 gramos. Los recipientes para las muestras no se llenarán menos de dos tercios ni más de nueve décimos de su capacidad. Inmediatamente después de cerrados serán envueltos en papel y almacenados en sitio oscuro.
c) Mantequilla en recipientes autorizados de peso unitario inferior a 0,25 kilogramos:
Se tomará como muestra el número de unidades enteras necesarias para conseguir un peso mínimo de la muestra de 200 gramos. El número de unidades enteras se tomarán con su envase y en este caso, aparte de los recipientes autorizados, se podrán emplear bolsas de plástico, protegidas por una bolsa de papel. Estas bolsas no se llenarán menos de dos tercios ni más de nueve décimos de su capacidad útil.
1.3 Conservación de las muestras.
No se adicionarán conservadores de ningún tipo a las distintas muestras de mantequilla, que se almacenarán en cámara fría.
Para análisis microbiológicos o examen organoléptico se conservarán las muestras a temperaturas comprendidas entre 0º C y 5º C.
1.4 Transporte de las muestras.
Las muestras serán transportadas al laboratorio lo más rápidamente posible después de la toma de muestras a temperaturas inferiores a + 10° C. Para análisis microbiológicos las temperaturas no deben sobrepasar los + 5º C.
1.5 Preparación de la muestra para las distintas determinaciones.
En los apartados a) y b) del punto 1.2 se colocará el recipiente con la muestra al baño María, sin sobrepasar la temperatura de + 39° C, hasta obtener una consistencia óptima y fluidez homogénea. La emulsión queda intacta, pero fluida, notándose claramente el nivel. Sacar del baño María y dejar reposar hasta enfriamiento.
En el caso c) del punto 1.2, una vez abiertos los paquetes que constituyen la muestra, se pasa con una espátula o cuchillo la mantequilla a un recipiente de boca ancha, que se colocará a continuación a baño María, sin sobrepasar la temperatura de + 39° C, hasta obtener una consistencia óptima y fluidez homogénea. La emulsión queda intacta, pero fluida, notándose claramente el nivel. Sacar del baño María y dejar reposar hasta enfriamiento.
2. DETERMINACIONES ANALITICAS
2.1 Indice de acidez de la grasa.
2.1.1 Principio.
El índice de acidez de la materia grasa en la mantequilla es el número de miligramos de hidróxido de potasio que se necesitan para neutralizar un gramo de materia grasa.
La materia grasa, después de separarla por fusión de la mantequilla, se disuelve en una mezcla de alcohol éter y luego se titula con una solución alcalina valorada.
2.1.2 Material y aparatos.
2.1.2.1 Balanza analítica.
2.1.2.2 Matraces Erlenmeyer de 300 mililitros.
2.1.2.3 Bureta graduada contrastada en divisiones de 0,1 mililitros.
2.1.3 Reactivos.
Los reactivos que se utilicen deben ser de calidad pura para análisis.
2.1.3.1 Solución alcohólica de hidróxido de potasio, 0,1 N valorada. Utilizar alcohol etílico absoluto.
2.1.3.2 Mezcla' de volúmenes iguales de alcohol etílico de 95-96 por 100 (v/v), o de alcohol desnaturalizado con metanol y de éter dietílico, neutro a la fenolftaleína.
2.1.3.3 Solución neutra de fenolftaleína al 1 por 100 (m/v) en alcohol etílico de 95-96 por 100 (v/v) o en alcohol etílico desnaturalizado con metanol.
Nota.–Alcohol desnaturalizado con metanol: 100 alcohol absoluto y cinco de metanol.
2.1.4 Procedimiento.
Para separar la materia grasa, fundir la muestra, dejarla reposar a 50-60° de dos a tres horas, decantar y filtrar con papel de filtro seco. Filtrar nuevamente, si el primer filtrado no está claro. Utilizar la materia grasa fundida, clarificada, bien mezclada.
En un matraz Erlenmeyer pesar, con precisión de un miligramo, 5-10 gramos de materia grasa. Añadir 50-100 ml. de la mezcla de alcohol etílico y éter dietílico y disolver en esta mezcla la materia grasa. Añadir 0,1 ml de la solución de fenolftaleína. Valorar con la solución alcalina hasta que aparezca una coloración rosa pálido que persista, al menos, diez segundos.
2.1.5 Cálculo.

ν = número de milímetros de la solución alcalina empleados.
t = normalidad de la solución alcalina.
 = masa en granos de la porción ensayada.
= masa en granos de la porción ensayada.
La diferencia entre los resultados de dos determinaciones paralelas no debe ser mayor de 0,1 mg. de hidróxido de potasio por un gramo de materia grasa.
2.1.6 Referencia.
FIL – IDE – GA – 1.969
2.2 Indice de refracción de la grasa.
2.2.1 Principio.
El índice de refracción de la materia grasa en la mantequilla es la razón entre la velocidad de una luz de longitud de onda determinada (luz de sodio) en el aire y la velocidad de esta misma luz en la materia grasa de la mantequilla a 40° C.
Mediante un refractómetro apropiado se determina el índice de refracción de la materia grasa obtenida por fusión de la mantequilla.
2.2.2 Material y aparatos.
2.2.2.1 Refractómetro provisto de escala graduada en índices de refracción, que permita efectuar lecturas hasta la tercera cifra decimal y cuyos prismas pueden calentarse mediante un líquido circulante, regulándose la temperatura termostáticamente con una aproximación de 7 0,1° C.
2.2.2.2 Luz de sodio; se puede utilizar también la luz blanca si el refractómetro tiene un dispositivo de compensación cromática.
2.2.3 Procedimiento.
Para separar la materia grasa fundir la muestra y dejarla reposar dos a tres horas a 50-60° C; decantar y filtrar con papel de filtro seco. Filtrar nuevamente si el primer filtrado no está claro. Utilizar la materia grasa fundida, clarificada, bien mezclada sin agua.
Preparar y regular el refractómetro según el modo de empleo del aparato. Ajustar la temperatura del líquido circulante a 40 = 0,1° C.
Colocar algunas gotas de materia grasa, preparada en la forma anteriormente descrita, entre los prismas del refractómetro, de manera que se llene por completo el espacio comprendido entre ellos. Dejar transcurrir algunos minutos para que la materia grasa alcance la temperatura de los prismas. Efectuar la lectura con cuatro cifras decimales. Corregir el índice de refracción obtenido añadiendo 0,000045 unidad de índice de acidez si este último (determinado según 1) es igual o superior a dos. Redondear la cuarta cifra decimal.
2.2.4 Cálculo.
La diferencia entre los resultados de determinaciones paralelas, no debe ser mayor de 0,0002 de la unidad de índice de refracción.
2.2.5 Referencias.
F. A. O. — B-5 — 1967
2.3 Cloruro sódico.
2.3.1 Principio.
Se entiende por contenido de sal (cloruro de sodio) de la mantequilla el porcentaje en masa de la sal (cloruro de sodio), determinado por el procedimiento que se describe a continuación.
Después de fundir la mantequilla añadiendo agua hirviendo, los cloruros de las mezclas se valoran con una solución de nitrato de plata, empleando cromato de potasio como indicador, según el procedimiento de Mohr, y se calcula el contenido de sal.,
2.3.2 Material y aparatos.
2.3.2.1. Balanza analítica.
2.3.2.2 Matraces Erlenmeyer, de 250 ml de capacidad.
2.3.2.3 Bureta graduada y contrastada en divisiones de 0,1 ml.
2.3.3 Reactivos.
2.3.3.1 Solución valorada de nitrato de plata, 0,1 N.
2.3.3.2 Solución de cromato de potasio al 5 por 100 (m/v) en agua destilada.
2.3.4. Procedimiento.
Ablandar la muestra en un recipiente cerrado, calentándola en baño de agua a la temperatura más baja posible, con objeto de no romper la emulsión. Frecuentemente es adecuada una temperatura comprendida entre 23 y 28° C, y en ningún caso la temperatura podrá exceder de 39° C. Agitar el recipiente que contiene la muestra a intervalos frecuentes durante el proceso de ablandamiento, con objeto de que la muestra se mezcle homogéneamente. Sacar el recipiente del baño de agua y agitarlo vigorosamente a intervalos frecuentes hasta que la muestra se haya enfriado, adquiriendo una consistencia espesa y cremosa. Esta operación puede realizarse con un agitador mecánico.
Efectuar una determinación en blanco, empleando los mismos reactivos, en las mismas cantidades y siguiendo el mismo procedimiento que se describe a continuación.
Pesar con una precisión de 10 mg., 5 gramos de muestra e introducirla en un matraz Erlenmeyer.
Añadir cuidadosamente 100 ml de agua destilada hirviendo. Dejar en reposo durante cinco-diez minutos, agitando por rotación de vez en cuando, mientras se enfría a una temperatura de 50/55° C. (temperatura de valoración). Añadir 2 ml. de solución de cromato de potasio. Mezclar agitando por rotación. Mientras se agita continuamente, valorar con la solución de nitrato de plata hasta que el cambio de color anaranjado pardo persista durante treinta segundos.
2.3.5. Cálculos.
El contenido de sal (expresado en porcentaje por masa de Na Cl.) es:

t = normalidad de la solución de nitrato de plata.
V1 = cantidad de ml. de solución de nitrato de plata, utilizados en la valoración.
V0 = cantidad de ml. de solución de nitrato de plata en el ensayo en blanco.
a = masa, en gramos, de la muestra utilizada. Redondear el resultado a 0,01 por 100.
La diferencia entre los resultados de dos determinaciones paralelas no deberá ser mayor de 0,02 gramos de cloruro de sodio por 100 gramos de producto.
Referencia:
FIL – 12 A – 1.969
2.4 Agua, extracto seco magro y grasa en una sola muestra.
2.4.1 Principio.
Se define el contenido de agua en la mantequilla como la pérdida de masa, expresado como porcentaje, en masa, según se determina por el procedimiento descrito.
Se define el contenido de extracto seco magro en la mantequilla como el porcentaje, en masa, de sustancias según se determina.
Se define el contenido de materia grasa en la mantequilla como el porcentaje, en masa, que se obtiene restando de 100 el contenido de agua y el del extracto seco magro.
El contenido de agua se determina gravimétricamente secando una cantidad conocida de mantequilla a + 102° C ± 2° C.
El contenido de extracto seco magro se determina gravimétricamente después de extraer con éter de petróleo la grasa de la mantequilla secada.
2.4.2 Material y aparatos.
2.4.2.1 Balanza analítica con una tolerancia de 0,1 mg.
2.4.2.2 Estufa de desecación, bien ventilada y controlada con termostato (ajustada para que funcione a una temperatura de 102° C ± 2° C.
2.4.2.3 Cápsulas metálicas, de porcelana o de vidrio, resistentes a la corrosión, que tengan por lo menos 25 milímetros de altura y 50 milímetros de diámetro.
2.4.2.4 Crisoles filtrantes de vidrio sinterizado (porosidad número 3) con matraces de filtración a la trompa.
2.4.2.5 Varilla con pieza final de material adecuado.
2.4.3 Reactivos.
Eter de petróleo con límites de ebullición entre + 30° C y + 60° C. Este reactivo no debe dejar ningún residuo por evaporación.
2.4.4 Procedimiento.
2.4.4.1 Preparación de la muestra.
Excepto cuando el mezclado no se considere necesario, la muestra debe mezclarse agitando con una varilla o con un agitador mecánico, lo más rápidamente posible, sin exceder de un minuto. La temperatura del mezclado deberá estar comprendida normalmente entre 23 y 28° C, pero en ningún caso deberá exceder de 35° C. Antes de pesar, la muestra deberá ponerse siempre a la temperatura ambiente.
2.4.4.2 Determinación de agua.
Secar la cápsula en la estufa hasta masa constante. Dejar enfriar la cápsula en desecador hasta la temperatura ambiente (treinta-treinta y cinco minutos) y pesar.
Pesar en la cápsula, de 5 a 10 gramos de la muestra de mantequilla. Mantener la cápsula en la estufa durante una hora, por lo menos. Dejar que se enfríe la cápsula en desecador a la temperatura ambiente (de treinta-treinta y cinco minutos) y pesar. Repetir el secado a intervalos de media hora hasta masa constante (variación igual o inferior a 0,5 mg.) Todas las pesadas se harán con una precisión de 0,1 mg. En el caso de que aumente la masa, se toma la masa mínima para el cálculo. No deben emplearse en esta determinación materiales absorbentes.
2.4.4.3 Determinación del extracto seco magro.
Secar el crisol de vidrio filtrante en la estufa hasta masa constante. Dejar enfriar el crisol a temperatura ambiente (treinta-treinta y cinco minutos) y pesar. Añadir de 10 a 15 ml. de éter de petróleo caliente a la cápsula, que contiene el extracto seco procedente de la determinación de agua, de manera que se disuelva la grasa.
Separar la mayor cantidad posible del sedimento adherido a la cápsula utilizando una varilla y pasar cuantitativamente la solución sobre la punta de la varilla al crisol. Repetir las operaciones cinco veces. Lavar el sedimento que queda en el crisol con 25 ml. de éter de petróleo caliente. Secar la cápsula y el crisol en la estufa durante dos horas. Dejar que la cápsula y el crisol se enfríen a la temperatura ambiente (treinta-treinta y cinco minutos) y pesar. Repetir las operaciones durante períodos de treinta minutos a la temperatura de secado hasta que la masa no disminuya más. Todas las pesadas se harán con una precisión de 0,1 mg.
2.4.5 Cálculos.
2.4.5.1 Método de cálculo de contenido de agua. Emplear la fórmula:

Siendo:
M = masa, en gramos de la muestra.
n = masa, en gramos de la muestra después de secar.
2.4.5.2 Método de cálculo del extracto seco magro. Emplear la fórmula:

Siendo:
A1 = masa, en gramos, del crisol vacío.
A2 = masa, en gramos, del crisol conteniendo sedimento.
B1 = masa, en gramos, de la cápsula vacía.
B2 = masa, en gramos, de la cápsula con sedimento residual.
M = masa, en gramos, de la muestra.
2.4.5.3 Método de cálculo del contenido de grasa.
Grasa % = 100 — (E + S).
E = porcentaje, en masa, de agua (calculada en 2.4.5.1).
S = porcentaje, en masa, de extracto seco magro (calculado en 2.4.5.2).
Para la determinación del contenido de agua, la diferencia entre el resultado de determinaciones paralelas no deberá exceder de 0,1 gramo de agua para 100 gramos de mantequilla.
Para la determinación del contenido de extracto seco magro la diferencia entre resultados de determinaciones paralelas no deberá exceder de 0,05 gramos de extracto seco magro para 100 gramos de mantequilla.
2.5 Detección de grasa vegetal en grasa de leche por cromatografía de gases de esteroles.
2.5.1 Principio.
Los digitónidos de esterol se disuelven en una mezcla de formamida y dimetil formamida, y los esteroles liberados se extraen con pentano y se separan por cromatografía de gases. Si se obtiene en el cromatograma un pico con el tiempo de retención del beta-sitosterol, se concluye la presencia de grasa vegetal en la muestra de grasa examinada
2.5.2 Material y aparatos.
2.5.2.1 Cromatógrafo de gases, equipado de un detector de inoización de llama, con un inyector de plata o de vidrio, con un sistema de inyección directa en la columna y con un registrador.
2.5.2.2 Columna de cromatografía de gases de vidrio o de acero inoxidable, de forma de U o en espiral, de uno a dos metros de longitud, diámetro interior 3-4 milímetros. Se recomienda el vidrio, pues algunos tipos de acero inoxidable dan resultados erróneos por alteración de los esteroles.
2.5.2.3 Microjeringa, capaz de proporcionar dosis de 5 ó 10 microlitros.
2.5.3 Reactivos.
2.5.3.1 Mezcla de volúmenes iguales de formamida y dimetil formamida.
2.5.3.2 n — pentano.
2.5.3.3 Relleno de la columna: fase estacionaria de goma de silicona (tipo metílico), estable hasta por lo menos 300° C que impregna, al 4 por 100, una tierra de diatomeas calcinada, lavada al ácido y silanizada, de granulometría 80/100 ó 100/120 de malla.
2.5.3.4 Solución para el ensayo de sensibilidad: 1 mg. de colesterol de 1 ml. de n-pentano, recientemente preparado a partir de la grasa de leche como se describe en (2.5.4.2).
2.5.3.5 Solución para el ensayo de resolución de los picos: 0,9 mg. de fitosterol de aceite de colza y 0,1 mg. de colesterol en 1 ml. de n-pentano. Los esteroles deben estar recientemente preparados según el procedimiento descrito en (2 5.4.2).
2.5.3.6 Solución para el ensayo de referencia: 1 mg. de fitosterol de aceite de soja en 1 ml. de n-pentano, recientemente preparado según se describe en (2.5.4.2).
2.5.3.7 Gas portador, nitrógeno.
2.5.3.8 Hidrógeno.
2.5.3.9 Oxígeno o aire.
2.5.4 Procedimiento.
2.5.4.1 Preparación de la muestra.
Fundir, aproximadamente, 50 gramos de la muestra de mantequilla en una estufa corriente a temperatura inferior a 50° C hasta separación de las fases acuosa y grasa. Separar la capa grasa por decantación y clarificar la grasa en una estufa a una temperatura de aproximadamente 40° C, filtrando sobre un papel de filtro seco, y evitando que pase la fase acuosa sobre el filtro.
2.5.4.2. Preparación de esteroles.
Posar en un matraz Erlenmeyer de 500 ml., aproximadamente, 15 gramos de materia grasa con una precisión de 0,1 gramos. Añadir 10 ml. de la solución de hidróxido de potasio y 20 ml. de etanol (95-96 por 100 v/v). Colocar sobre el matraz Erlenmeyer el refrigerante de aire, calentar en baño de agua hirviendo, agitando por rotación, hasta que la solución se haga clara, y continuar la ebullición media hora más.
Añadir 60 ml. de agua y luego 180 ml. de etanol (95-96 por 100 v/v) y llevar la temperatura a aproximadamente 40° C. Añadir 30 ml. de la solución alcohólica de digitonina (1 por 100), agitar y dejar enfriar. Colocar el matraz en un refrigerador regulado a 5º C, aproximadamente, durante doce horas o una noche.
Recoger el precipitado del digitónido de esterol por filtración sobre un papel de filtro de velocidad media en un embudo Buchner (8 centímetros de diámetro). Lavar el precipitado con agua aproximadamente a 5ºC hasta que el filtrado no forme espuma, luego lavar una vez con 25-50 ml. de etanol (95-96 por 100 v/v) y después una vez con 25-50 ml. de éter dietílico.
Desecar el papel de filtro con el precipitado en un vidrio de reloj en estufa a 102 + 2° C durante diez o quince minutos. Separar el precipitado en forma de película, plegando en dos partes el papel de filtro.
Disolver en un pequeño tubo de ensayo aproximadamente 10 mg. de digitónido de esterol en 0,5 ml. de una mezcla de volúmenes iguales de formamida y dimetil formamida. Calentar ligeramente, si es necesario. Después enfriar, añadir 2,5 ml. de n-pentano y agitar. Dejar reposar hasta que la separación entre las capas sea neta, y usar la capa superior, que contiene los esteroles liberados para el análisis cromatográfico.
2.5.4.3 Condiciones de la cromatografía de gases.
Temperatura de la columna: 220-250° C. Temperatura del sistema de inyección, si puede calentarse por separado: 20-40° C por encima de la temperatura de la columna. Gasto de nitrógeno: 30-60 ml/min. Desconectar el detector y equilibrar las columnas nuevas en estas condiciones durante dieciséis-veinticuatro horas. Conectar el detector, encender la llama y regular el gasto de hidrógeno y oxígeno o aire para obtener una altura de llama y una sensibilidad adecuada. Poner en marcha el registrador y dejar que el papel se desenrolle a una velocidad adecuada, ajustar el cero y el atenuador. Si la línea de base es estable, el aparato está listo para usarse.
2.5.4.4 Ensayo de sensibilidad.
Inyectar tres a cinco microlitros de la solución para el ensayo de sensibilidad (2.5.3.4). Sólo aparecerá un pico de colesterol en el cromatograma (figura I). Ajustar el atenuador de modo que se utilice aproximadamente toda la escala del registrador.
ESTEROLES DE LA MATERIA GRASA DE LA LECHE
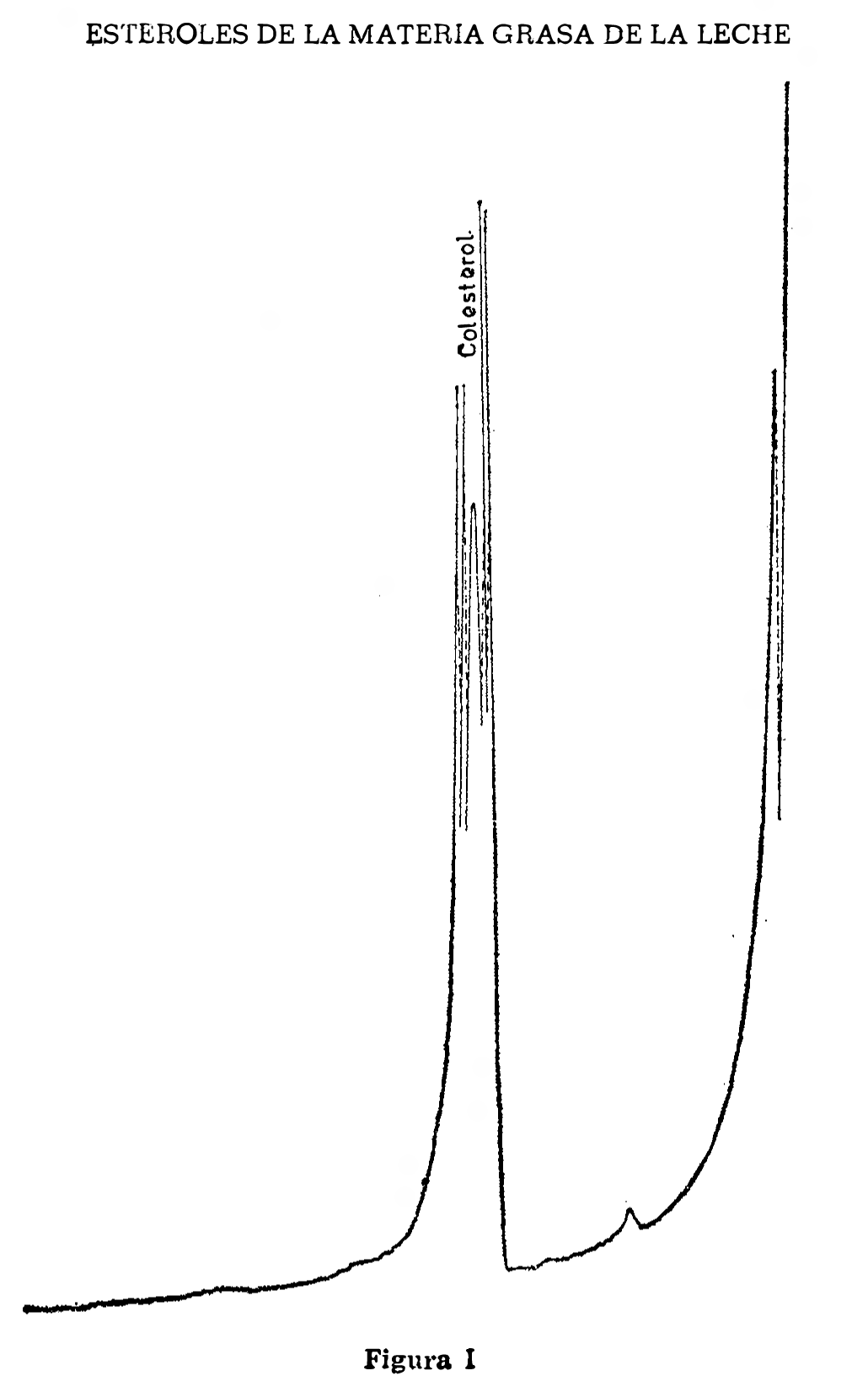
2.5.4.5 Ensayo de resolución de los picos.
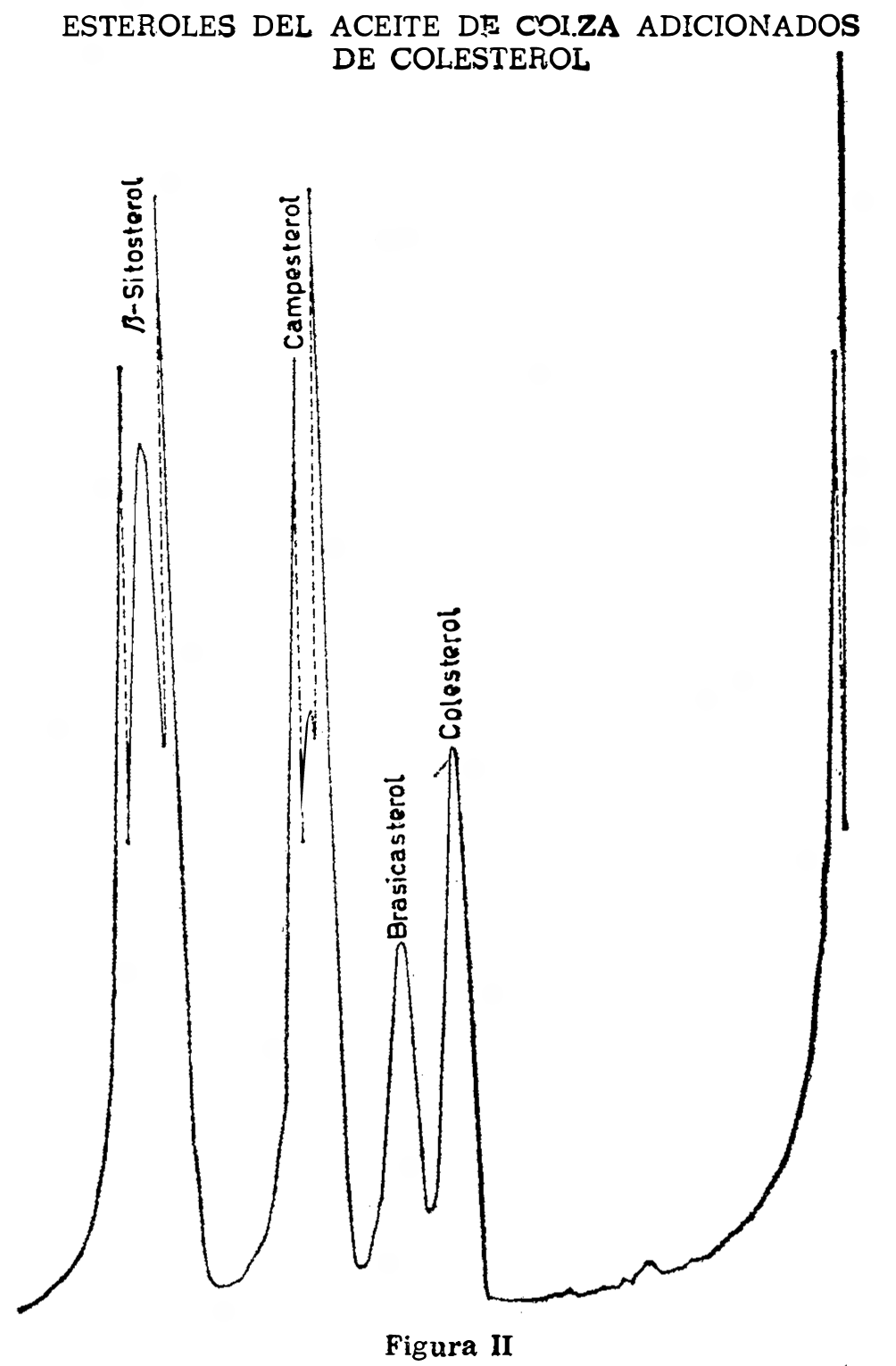
Inyectar tres a cinco microlitros de la solución para el ensayo de resolución de los picos (2.5.3.5). Aparecerán en el cromatograma los picos de colesterol, brasicosterol, camposterol y. beta-sitosterol. (Fig. II). Medir las distancias de retención (distancia desde el punto de inyección hasta el punto de altura máxima del pico) de los picos, dCH para el colesterol, dB para el brasicosterol, dC para el camposterol y dS para el beta-sitosterol y las anchuras de la base de los picos (dimensión de retención entre las intersecciones con la línea de base de las tangentes en los puntos de inflexión situados en la parte anterior y posterior del pico) para el colesterol y para el brasicosterol. La resolución de los picos, expresados por la fórmula:
PR = 2 (dB — dCH)/(WB + WCH)
debe ser por lo menos igual a 1.
Calcular los tiempos de retención relativos (colesterol 1,00) para el brasicosterol, el camposterol y el beta-sitosterol.
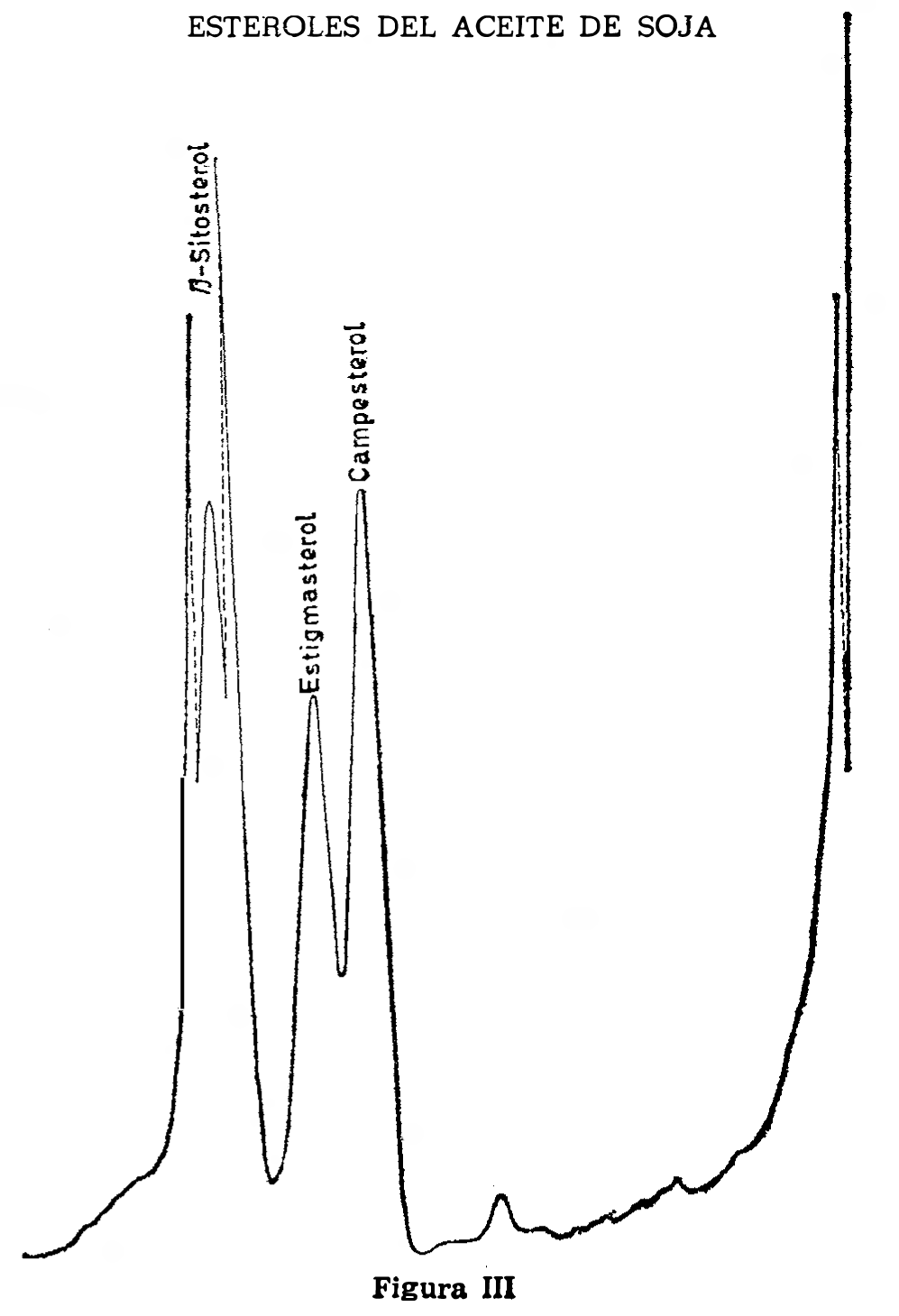
2.5.4.6 Ensayo de referencia.
Inyectar tres a cinco microlitros de la solución para el ensayo de referencia (2.5.3.6). Los picos de camposterol, estigmasterol y de beta-sitosterol deben aparecer sobre el cromatograma (Fig. III). Medir las distancias de retención de los picos, dC para el camposterol, dST para el estigmasterol y dS para el beta-sitosterol.
Calcular los tiempos de retención relativos que son aproximadamente.
Colesterol: 1,00. (Aproximadamente quince minutos.)
Brasicosterol: 1,13 - 1,15.
Camposterof: 1,32 - 1,34.
Estigmasterol: 1,44 - 1,46.
Beta-sitosterol: 1,66 - 1,68.
2.5.4.7 Análisis.
Inyectar tres a cinco microlitros de la solución a analizar y dar al botón del atenuador hasta obtener un factor de atenuación cuatro veces inferior (se obtiene generalmente en dos pases del botón). Registrar el cromatograma.
2.5.5 Cálculo.
Si en el cromatograma, un pico tiene un tiempo de retención relativo igual al de beta-sitosterol, y una altura, que corresponde al menos a un 2 por 100 de la escala, se concluye la presencia de beta-sitosterol y la muestra de grasa examinada, a partir de la cual se han aislado los esteroles, se considera que contiene grasa vegetal. La presencia en el cromatograma de picos de otros fitosteroles, como el camposterol o el estigmasterol, refuerza esta conclusión.
Por este método se puede demostrar la presencia de al menos un 0,5 por 100 de beta-sitosterol en la mezcla de esteroles. El límite de detección de la grasa vegetal en la grasa de leche no se puede indicar, porque depende del contenido en betasitosterol de la grasa añadida, es decir, de la naturaleza de esta grasa o de la mezcla de grasas añadidas a la grasa de leche.
2.5.6 Referencias.
1. Norma internacional FIL IDF 54: 1970.
2.6 Fosfatasa residual en mantequilla.
2.6.1 Principio.
El ensayo se basa en la acción del enzima fosfatasa sobre el sustrato fenil fosfato disódico, con liberación de fenol y fosfato. La cantidad de fenol liberada se determina por adición de un reactivo que da color azul en presencia de fenol.
Cantidades superiores a dos equivalentes de fenol en 0,5 gramos de mantequilla indican una pasteurización insuficiente.
2.6.2 Material y aparatos.
2.6.2.1 Cuchillo o espátula de acero inoxidable.
2.6.2.2 Baño de agua a 37-38º C.
2.6.2.3 Termómetro.
2.6.2.4 Pipetas un ml.
2.6.2.5 Embudo de cinco cm. de diámetro.
2.6.2.6 Papel Whatman No. 42 o No. 2.
2.6.2.7 Tubos graduados a 5,0 y 10,0 ml.
2.6.2.8 Fotómetro con filtro de transmitancia máxima a 610 m/u.
2.6.2.9 Centrífuga.
2.6.2.10 Pera de goma para pipetar.
2.6.3 Reactivos.
2.6.3.1 Tampón hidróxido-borato de bario: Disolver 18 gramos de Ba (OH)2 8 H2O y 8 g. de H3. BO3 en agua y diluir a un litro con agua.
2.6.3.2 Tampón de desarrollo de color: PH 9,8 ± 0,15 a 25° C. Disolver 6,0 g. de metaborato sódico (Na BO2) y 20 gramos Na C1 en agua y diluir a un litro con agua.
2.6.3.3 Tampón de dilución de color: Diluir 100 ml. de tampón de desarrollo de color a un litro con agua.
2.6.3.4 Sustrato tampón: Disolver 0,10 g. de fenil fosfato disódico cristalino libre de fenol en 100 ml. de tampón hidróxido-borato de bario (2.6.3.1). (Los cristales de Na2 C6 H5, PO1 deben guardarse en congelador o en desecador). Si Na2 C6 H5, PO1 no está libre de fenol, purificado como sigue: Disolver 0,5 g. en 4,5 ml. de agua, añadir 0,5 ml. de tampón hidróxido-borato de bario (2.6.3.1) y dos gotas de reactivo BQC (2.6.3.5) y dejar reposar treinta minutos. Extraer el color con 2,5 ml. de alcohol butílico (2.6.3.7) y dejar reposar hasta que el alcohol se separe. Retirar el alcohol con un cuentagotas y desecharlo. Diluir 1,0 ml. de la solución acuosa a 100 ml. de tampón hidróxido-borato de bario (2.6.3.1). Calentar la solución a 85° C dos minutos, tapar inmediatamente y conservar en refrigerador. La solución es estable un año si las porciones son recogidas con mínima exposición a la atmósfera,
2.6.3.5 Solución BQC (s, 6-d ibromoquinona cloroimida) o reactivo de Gibbs: Disolver 40 mg. BQC en polvo en 100 ml. de alcohol absoluto o metanol y pasarlo a un frasco cuentagotas oscuro. El reactivo permanece estable por lo menos un mes, si se guarda en congelador; no usarlo después de que empiece a ponerse pardo. Guardar BQC en polvo en congelador o en desecador. Comprobar los nuevos lotes de BQC antes de usarlo, preparando una curva patrón con fenol y comparando la curva obtenida con la de BQC, que se sabe es satisfactorio. Repetir la prueba, al menos, semestralmente.
2.6.3.6 Solución de sulfato de cobre para los patrones: Disolver 0,05 g. Cu SO4 5H2O en agua y diluir a 100 ml.
2.6.3.7 Alcohol butílico: Usar alcohol butílico normal, punto de ebullición 116-118° C. Para ajustar el pH| mezclar un litro con 50 ml. de tampón de desarrollo de color. Guardar en recipiente con tapón de vidrio.
2.6.3.8 Soluciones patrón de fenol:
2.6.3.8.1 Solución madre. Pesar exactamente un gramo de fenol puro, llevarlo a un matraz aforado de un litro, diluirlo con agua hasta un litro y mezclar (1 ml. = 1 mg. de fenol). (La solución es estable varios meses en refrigerador.)
2.6.3.8.2 Patrones de trabajo. Diluir 10 ml. de la solución madre con agua hasta un litro y mezclar (1 ml. = 10/mg. 0,00001 g. o 10 unidades de fenol). Usar esta solución patrón para preparar soluciones patrón más diluidas: Por ejemplo, diluir 5, 10, 30 y 50 ml. con agua hasta 100 ml. para preparar soluciones patrón que contengan 0,5, 1, 3 y 5 mg. o unidades de fenol/ml., respectivamente. Guardar estas soluciones patrón en refrigerador no más de una semana.
Análogamente, preparar a partir de la solución madre (2.6.3.8,1) soluciones patrón que contengan 20, 30 y 40 unidades/ml.
Medir las cantidades adecuadas de las soluciones patrón de trabajo en una serie de tubos (preferiblemente graduados a 5 y 10 ml.) para conseguir un intervalo adecuado de patrones, según se necesite, que contengan 0 (control o prueba en blanco), 0,5, 1, 3, 5, 10, 20. 30 y 40 unidades. Para aumentar el brillo de las soluciones azules y mejorar la estabilidad de los patrones, añadir 1 ml. de solución de Cu SO4 (2.6.3.6) a cada tubo. Luego añadir 5 ml. de tampón de dilución de color (2.6.3.3) y diluir con agua hasta 10 ml., añadir cuatro gotas (0,03 ml.) de la solución BQC (2.6.3.5), mezclar y dejar desarrollar el color azul treinta minutos a temperatura ambiente.
Leer las intensidades de color en el fotómetro con filtro de 610 mm., restar el valor de la prueba en blanco del valor de cada patrón de fenol y preparar la curva patrón (debe ser una línea recta).
Si los patrones han de usarse para comparación visual, guardar el refrigerador. Preparar semanalmente una serie nueva.
2.6.4 Procedimiento.
Tomar la muestra por debajo de la superficie con cuchillo y espátula limpios y proceder como sigue:
Pesar 1 g. de muestra (preferiblemente por duplicado) sobre un pedazo de papel encerado de aproximadamente una pulgada cuadrada e introducir el papel con la muestra dentro del tubo. Análogamente, pesar otra muestra y colocarla en un tubo como control o patrón.
Calentar el patrón aproximadamente un minuto a 85-90° en vaso de agua hirviendo (cubierto así el tubo entero se calienta a 85-90°) y se enfría a temperatura ambiente. A partir de este momento tratar en la misma forma el patrón y el problema.
Añadir 10 ml. de sustrato patrón (2.6.3.4). Tapar el tubo y mezclar. Inmediatamente después de añadir el sustrato, incubar una hora en baño de agua a 37-38°, mezclando o agitando el contenido de vez en cuando.
Calentar en vaso de agua hirviendo casi un minuto, calentando hasta 85-90° (utilizar termómetro en otro tubo del mismo tamaño y forma que contenga el mismo volumen de líquido) y enfriar a temperatura ambiente en recipiente de agua fría.
Pipetar en un ml. de solución de Zn SO4 H2O de 6,0 gramos/ 100 ml. y mezclar por completo (el pH de la mezcla debe ser de 9,0-9,1.
Filtrar (se recomienda embudo de cinco centímetros y papel Whatman número 2) y recoger 5,0 mi. de filtrado en el tubo, preferentemente graduado a 5,0 y 10,0 ml.
Añadir 5,0 ml. de tampón de desarrollo de color (2.6.3.2) (el pH de la mezcla debe ser 9,3-9,4).
Añadir cuatro gotas de la solución BQC (2.6.3.5) y dejar treinta minutos a temperatura ambiente para desarrollo de color (para detectar únicamente la pasteurización, añadir solamente dos gotas de solución BQC).
Determinar la identidad del color azul por uno de los siguientes métodos:
a) Con fotómetro.–Leer intensidades de color de soluciones en blanco y problema (utilizando filtro con transmitancia máxima a aproximadamente 610 milímetros), restar la lectura de la prueba en blanco de la del problema y expresar el resultado en equivalentes fenol mediante referencias a la curva patrón, obtenida con las correspondientes soluciones (2.6.3.8.2). Generalmente es innecesaria la extracción con alcohol butílico cuando se utiliza el fotómetro; si se hace la extracción con alcohol butílico cómo en b), centrifugar la muestra cinco minutos para romper la emulsión y separar el agua suspendida en la capa de alcohol. (Para esta finalidad puede, adaptarse una centrífuga Babcock haciendo adaptadores especiales para tubos en la forma siguiente: Cortar una sección de un cuarto de grueso de un tapón de goma de diámetro adecuado que ajuste en el fondo del vaso de centrifugación.) Pegar dos tapones de corcho de diámetro adecuado, perforar en el centro un orificio de dimensiones adecuadas para alojar un tubo ajustadamente, e introducir la sección doble de corcho en el vaso. Después-de centrifugar, quitar casi todo el alcohol butílico con pipeta provista de pera de goma en el extremo superior. Filtrar dentro de la cubeta del fotómetro y leer con filtro, cuyo máximo de transmitancia es aproximadamente de 650 milímetros.
b) Con patrones visuales.–Con muestras que producen más de cinco unidades, comparar colores en tubos con los de patrones de fenol en Solución acuosa (2.6,3.8.2). Para cuantificar resultados en los casos dudosos (p. e. problemas que producen 0,5-5 unidades de color), extraer con alcohol butílico (2.6.3.7). Añadir 5,0 ml. de alcohol (2.6.3.7) e invertir el tubo lentamente varias veces; centrifugar como en a) si es necesario incrementar la transparencia de la capa de alcohol, y comparar el color azul con los colores de los patrones de fenol (2.6.3.8.2), análogamente tratados.
En los problemas que se consideran muy positivos durante el desarrollo de color (p. e. 20 unidades), en los que cuatro gotas de solución de BQC (2.6.3.5) pueden ser insuficientes para combinar con todo el fenol, pipetar proporción adecuada de contenidos dentro de otro tubo, diluir hasta 10,0 ml. con tampón de dilución de color (2.6.3.3), y añadir dos gotas adicionales de solución BQC (2.6.3.5). Con cada problema, diluir y tratar la prueba en blanco análogamente. Si la prueba sobre la muestra diluida es todavía muy fuertemente positiva, diluir de nuevo en la misma forma hasta que el color final esté dentro del intervalo de los patrones visuales o de la curva patrón del fotómetro. Dejar treinta minutos para el desarrollo de color después de la última adición de la solución de BQC (2.6.3.5) antes de hacer la lectura final. Para corregir lecturas por dilución, multiplicar por 2 para dilución 5 +.5, por. 10 para dilución 1 + 9 y por 50 dilución 1 + 9, seguida por dilución 2 + 8, etc.
2.6.5 Cálculo.
Cuando se utilice 1,0 gramos de mantequilla y se añadan 11,0 ml. de líquido, multiplicar el valor de la lectura por 1,1 para convertir el resultado en equivalentes de fenol/0,5 gramos de mantequilla. (Valores mayores de dos equivalentes/0,5 gramos de mantequilla indican pasteurización insuficiente.)
2.6.6 Referencias.
1. A. O. A. C. Official Methods of analysis, 11 Ed. (1970).
2.7. Indices de ácidos grasos volátiles solubles e insolubles.
2.7.1. Principio.
El índice de ácidos grasos volátiles solubles (índice de Reichert o Reicher-Meissl-Wollny) es el número de milímetros de una solución acuosa de álcali 0,1 N, requerido para neutralizar los ácidos grasos volátiles solubles en agua de cinco gramos de grasa en las condiciones que se especifican.
El índice de ácidos grasos volátiles insolubles (índice de Polenske) es el número de mililitros de polución acuosa de álcali 0,1 N requerido para neutralizar los ácidos grasos volátiles insolubles en agua, obtenidos de cinco gramos de grasa, en las condiciones especificadas en el método.
Después de saponificar la grasa con una solución de hidróxido sódico en glicerina, la solución jabonosa se diluye con agua y se acidifica con ácido sulfúrico. Los ácidos grasos volátiles se destilan y los ácidos grasos insolubles se separan de los solubles por filtración. La solución acuosa de ácidos solubles y la solución etanólica de ácidos insolubles se valoran separadamente con una solución de álcali normalizada.
El método es empírico, porque sólo determina una parte de estos ácidos. Por tanto, las especificaciones referentes al procedimiento y aparatos se deben seguir rigurosamente para obtener resultados exactos y reproducibles.
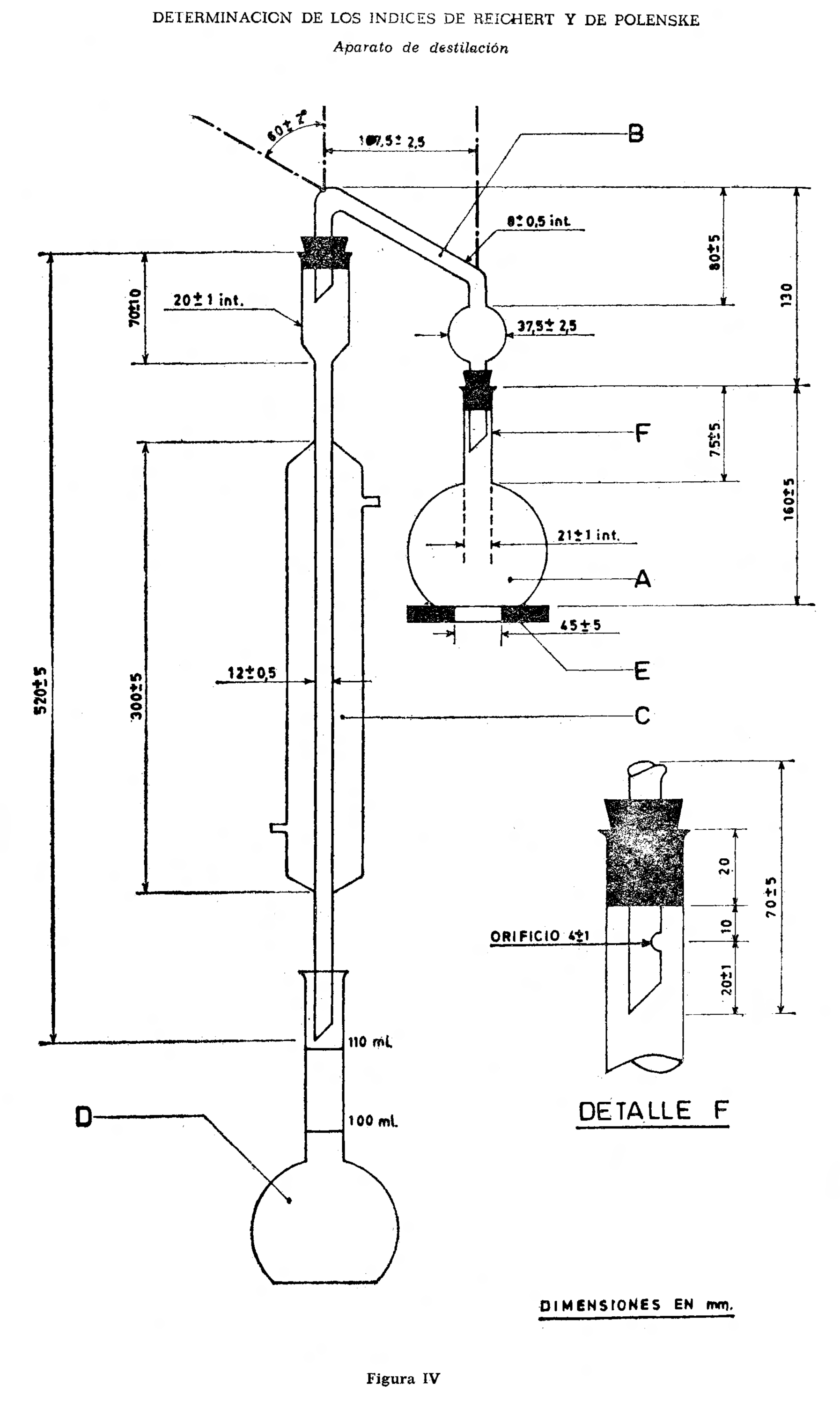
2.7.2 Material y aparatos. (Figura IV.)
2.7.2.1 Matraz de fondo plano de vidrio al borosilicato de 300 ml. de capacidad (A).
2.7.2.2 Cabeza de destilación (B).
2.7.2.3 Refrigerante (C).
2.7.2.4 Receptor, que consiste en un matraz aforado con las rayas circulares de aforo a 100 y 110. ml. (D).
2.7.2.5 Lámina de asbesto de 120 milímetros de diámetro, seis milímetros de espesor con una abertura central circular de 40 a 50 milímetros de diámetro, para sostener el matraz durante el calentamiento (E).
2.7.2.6 Piedra pómez triturada que pasa a través de un tamiz de malla circular de dos milímetros y son retenidos en un tamiz de malla circular de 1,44 milímetros, según el proyecto de Recomendación ISO No. 805.
En la figura se representan las dimensiones en milímetros y el montaje del aparato de destilación. Para las conexiones se pueden utilizar tapones de caucho, neopreno o silicona, o juntas de vidrio esmerilado standard 24/40.
2.7.3 Reactivos.
Todos los reactivos deben ser puros para análisis.
2.7.3.1 Glicerina (d = 1,26; 98 por 100 p/p).
2.7.3.2 Solución acuosa de hidróxido sódico (44 por 100 p/p), conservada en botella protegida del dióxido de carbono. Usar la porción límpida libre de precipitado de carbonatos.
2.7.3.3 Agua destilada, hervida durante quince minutos para eliminar el dióxido de carbono.
2.7.3.4 Solución de ácido sulfúrico 1 N.
2.7.3.5 Solución acuosa de hidróxido sódico o potásico 0,1 N, exactamente normalizada.
2.7.3.6 Solución indicadera de fenolftaleína (1 por 100 en etanol de 95-96 por 100).
2.7.3.7 Etanol, (95-96 por 100 v/v), neutro a la fenolftaleína.
El agua usada debe ser destilada o de una pureza, por lo menos, equivalente.
2.7.4 Procedimiento.
2.7.4.1 Preparación de la muestra.
Como en (2.5.4.1).
2.7.4.2 Determinación del índice de ácidos grasos volátiles solubles.
Pesar 5 + 0,01 gramos de grasa en el matraz A. Añadir 20 gramos (16 ml.) de glicerina y dos ml. de solución de hidróxido sódico (44 por 100). Para añadir la solución de hidróxido sódico, usar una bureta protegida de la entrada de dióxido de carbono y limpiar la punta de la bureta desechando las primeras gotas. Calentar el matraz a fuego-directo, evitando sobrecalentar y agitando continuamente, hasta que el líquido no forme más espuma y se vuelva límpido. Dejar enfriar el matraz hasta 90° C. Añadir 90 ml. de agua destilada recientemente hervida, a la misma temperatura aproximadamente, y mezclar. El líquido debe quedar límpido. Añadir de 0,6 a 0,7 gramos de piedra pómez y, después, 50 ml. de solución de ácido sulfúrico 1 N. Conectar inmediatamente el matraz al aparato de destilación y calentarlo, ligeramente hasta que los ácidos grasos libres formen una capa superficial límpida. Empezar a calentar y regular la llama de modo que se recojan en el matraz aforado 110 ml. de destilado en diecinueve-veintiún minutos, tomando como principio del período de destilación el momento en que se forma la primera gota en el refrigerante. Regular el flujo de agua del refrigerante de modo que se mantenga la temperatura del agua que sale del refrigerante a 20 + 1º D.
Cuando se hayan recogido exactamente 110 ml. de destilado, quitar el mechero inmediatamente y sustituir el matraz aforado por un pequeño vaso. Mezclar el contenido del matraz aforado agitando suavemente y sumergir el matraz en un baño de agua a 20 + 1º C durante diez-quince minutos, estando la señal de 110 ml. del matraz aforado a centímetro por debajo del nivel del agua del baño. Agitar el matraz de cuando en cuando. Tapar el matraz y mezclar invirtiéndolo cuatro o cinco veces, sin agitar. Filtrar los 110 ml. de destilado por un papel filtro seco de velocidad media (diámetro 80-90 milímetros), que se ajusta cómodamente en un embudo. El filtrado debe ser límpido. (El filtro debe ser de tales dimensiones, que un volumen de 15 ml. lo llene completamente). Pipetar 100 ml. de filtrado y pasarlos a un matraz Erlenmeyer de 300 ml., añadir 0,5 ml. de la solución indicadora de fenolftaleína y valorar con la solución acuosa de álcali standard 0,1 N hasta un color rosa persistente durante cero-cinco-un minutos.
2.7.4.3 Ensayo en blanco.
Hacer un ensayo en blanco sin grasa y, en lugar de saponificar a fuego directo, calentar en baño de agua hirviendo durante quince minutos.
No se requerirán para la valoración más de 0,5 ml. de la solución de álcali normalizada. En otro caso, se deben preparar nuevas soluciones del reactivo.
2.7.4.4 Determinación del índice de ácidos grados volátiles insolubles. (Polenske.)
Lavar el filtro con tres porciones sucesivas de 15 ml. de agua destilada, a la temperatura de 20 + 1º C, habiendo pasado previamente cada una a través del refrigerante del vaso pequeño y del matraz aforado. Poner el embudo y el filtro en el cuello de un matraz cónico, limpio y seco, de 200 ml. de capacidad. Disolver los ácidos grasos insolubles repitiendo los lavados, usando ahora porciones de 15 ml. de etanol (95-96 por 100 v/v). Valorar con la solución acuosa del álcali normalizada (0,1 N) el conjunto de los lavados con etanol usando 0,5 ml. de solución indicadora de fenolftaleína, hasta un color rosa persistente durante 0,5-1 minutos.
2.7.5 Cálculo.
2.7.5.1 Indice de ácidos grasos volátiles solubles (índice de Reichert).
Indice de Reichert = 11 t (ν1 — b)
Siendo:
ν1 = Volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en la valoración de la muestra.
b = Volumen en mililitros de la solución normalizada (0,1 N) de álcali, utilizados en el ensayo en blanco.
t = Normalidad exacta de la solución normalizada (0,1 N) de álcali.
Redondear el resultado a la primera cifra decimal.
2.7.5.2 Indice de ácidos grasos volátiles insolubles (índice de Polenske).
Indice de Polenske = 10 t ν2
ν2 = Volumen en mililitros de la solución normalizada (0,1 N) de álcali utilizada en la valoración de la muestra.
t = Normalidad exacta de la solución normalizada (0,1 N) de álcali.
Redondear el resultado a la primera cifra decimal.
2.7.5.3 Reproducibilidad de resultados.
La diferencia entre los resultados de dos determinaciones duplicadas (resultados obtenidos simultáneamente o inmediatamente uno detrás de otro por el mismo analista) no debe exceder de 0,5 para el índice de Reichert o de 0,3 para el índice de Polenske.
2.7.6 Referencias.
Norma internacional FIL-IDF 37: 1966.
2.8 Indice de Kirschner.
2.8.1 Método operatorio.
1. Neutralizar 100 ml. del destilado Reichert-Meissl con solución de Ba (OH)2 0,1 N, con toda precisión, hasta lograr un débil color rosado, empleando 0,5 ml., del indicador. Realizar la titulación en un matraz cerrado para evitar la absorción de CO2
2. Añadir 0,3 gramos de Ag2 SO4, en forma de polvo fino. Dejar reposar la mezcla una hora, agitando con frecuencia y filtrándola después.
3. Recoger 100 ml. del filtrado, colocarlo en un frasco de destilación de 300 ml., añadir 35 ml. de agua destilada y 10 ml. de H2 SO4 diluído. Añadir un trozo de alambre de aluminio o varios trozos de piedra pómez para evitar que el líquido se rebose. Unase al destilador y comiéncese la destilación a la velocidad de 110 ml. en unos veinte minutos.
4. Después de recoger 110 ml. del destilado, filtrar esta cantidad total y titular 100 ml. con Na OH, 1 N, empleando 0,5 ml. del indicador hasta lograr un tono rosado que persista durante dos o tres minutos.
5. Preparar y realizar una prueba en blanco semejante a la anterior en todos sus aspectos.
2.8.2 Cálculo.

A = Titulación de la muestra — titulación en blanco.
B = ml. de Ba (OH)2 0,1 N, requeridos para neutralizar los 100 ml. originales del destilado de Reichert-Meissl.
2.8.3 Referencias.
Norma internacional AOCS 5-40.
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid




