Agencia Estatal Boletín Oficial del Estado






 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.Incluye la corrección de errores publicada en BOE núm. 42, de 18 de febrero de 2004. Ref. BOE-A-2004-2974
[Bloque 2: #preambulo]
La Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente, incorporó al ordenamiento jurídico español las normas sustantivas de la Directiva 98/81/CE del Consejo, de 26 de octubre de 1998, por la que se modifica la Directiva 90/219/CEE relativa a la utilización confinada de microorganismos modificados genéticamente, y de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, sobre la liberación intencional en el medio ambiente de organismos modificados genéticamente y por la que se deroga la Directiva 90/220/CEE del Consejo.
No obstante, tal y como pone de manifiesto en su exposición de motivos, la Ley 9/2003, de 25 de abril, efectúa una incorporación parcial al derecho español que se limita a recoger y adaptar aquellas normas de las directivas mencionadas que, en razón de su naturaleza jurídica y para su plena efectividad en el ordenamiento interno, se encuentran sometidas al principio de reserva de ley, en tanto que la incorporación de aquellas otras normas de las directivas de contenido técnico o de carácter coyuntural, no sujetas al principio citado, se han diferido al ámbito reglamentario.
De otra parte, desde la fecha de su entrada en vigor, las normas objeto de las directivas citadas se han visto afectadas por el ordenamiento de la Unión Europea desde dos perspectivas diferentes.
En primer lugar, por la publicación de diversas decisiones de la Comisión y del Consejo que complementan el contenido de ambas directivas, entre las que se pueden citar la Decisión 2000/608/CE de la Comisión, de 27 de septiembre de 2000, referente a las notas de orientación para la evaluación del riesgo descrita en el anexo II de la Directiva 90/219/CEE; la Decisión 2001/204/CE del Consejo, de 8 de marzo de 2001, por la que se completa la Directiva 90/219/CEE con respecto a los criterios por los que se establece la inocuidad de los microorganismos modificados genéticamente para la salud humana y el medio ambiente; la Decisión 2002/623/CE de la Comisión, de 24 de julio de 2002, por la que se establecen una notas de orientación complementarias al anexo II de la Directiva 2001/18/CE; la Decisión 2002/811/CE del Consejo, de 3 de octubre de 2002, por la que se establecen unas notas de orientación complementarias al anexo VII de la Directiva 2001/18/CE; la Decisión 2002/812/CE del Consejo, de 3 de octubre de 2002, por la que se establece, de conformidad con la Directiva 2001/18/CE, el modelo de resumen de la notificación de la puesta en el mercado de organismos modificados genéticamente como producto o componentes de producto; la Decisión 2002/813/CE del Consejo, de 3 de octubre de 2002, por la que se establece, de conformidad con la Directiva 2001/18/CE, el modelo de resumen de la notificación de la liberación intencional en el medio ambiente de organismos modificados genéticamente con fines distintos de su puesta en el mercado; la Decisión 2003/701/CE de la Comisión, de 29 de septiembre de 2003, por la que se establece un modelo de presentación de los resultados de la liberación intencional en el medio ambiente de plantas superiores con una finalidad distinta de la de su comercialización con arreglo a la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, y la Decisión de la Comisión por la que se establecen las disposiciones pormenorizadas de funcionamiento de los registros establecidos con arreglo a la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, para el registro de información relativa a las modificaciones genéticas en organismos modificados genéticamente.
Así mismo es preciso considerar las disposiciones contenidas en el Reglamento (CE) n.º 1946/2003 del Parlamento Europeo y del Consejo, de 15 de julio de 2003, relativo al movimiento transfronterizo de organismos modificados genéticamente, que completa el contenido de la citadas directivas, en el Reglamento (CE) n.º 1830/2003 del Parlamento Europeo y del Consejo, de 22 de septiembre de 2003, relativo a la trazabilidad y etiquetado de organismos modificados genéticamente y a la trazabilidad de los alimentos y piensos producidos a partir de estos y por el que se modifica la Directiva 2001/18/CE, y en el Reglamento (CE) n.º 1829/2003 del Parlamento Europeo y del Consejo, de 22 de septiembre de 2003, sobre alimentos y piensos modificados genéticamente, lo que obliga a recoger estas modificaciones en una norma de rango reglamentario.
Además, la efectiva aplicación de la Ley 9/2003, de 25 de abril, implica desarrollar reglamentariamente, entre otros, diversos aspectos de su articulado relacionados con la estructura, composición y funciones del Consejo interministerial de organismos modificados genéticamente y de la Comisión Nacional de Bioseguridad, con los requisitos para la realización de actividades de utilización confinada, liberación voluntaria con fines distintos a su comercialización y a la comercialización de organismos modificados genéticamente, con las normas sobre información, vigilancia y control de estas actividades, así como en materia de régimen sancionador.
De acuerdo con lo expuesto, es preciso proceder a la aprobación y publicación de una norma que permita al Gobierno desarrollar el contenido de la Ley 9/2003, de 25 de abril, y finalizar, a la par, el proceso de incorporación al ordenamiento español de las directivas y demás normas comunitarias anteriormente citadas; norma que, por su naturaleza jurídica y de conformidad con la habilitación objeto de la disposición final quinta de la citada ley, debe revestir el rango y forma de un real decreto.
En la elaboración de este real decreto han sido oídas las comunidades autónomas, así como los sectores afectados.
En su virtud, a propuesta de los Ministros de Medio Ambiente, de Agricultura, Pesca y Alimentación, de Sanidad y Consumo y de Ciencia Y Tecnología, con la aprobación previa de la Ministra de Administraciones Públicas, de acuerdo con el Consejo de Estado y previa deliberación del Consejo de Ministros en su reunión del día 30 de enero de 2004,
DISPONGO:
[Bloque 3: #aunico]
Se aprueba el Reglamento general para el desarrollo y ejecución de la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente, que se inserta a continuación de este real decreto.
[Bloque 4: #daprimera]
De conformidad con lo establecido en la disposición adicional tercera de la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente, se crea el Registro central de organismos modificados genéticamente, adscrito al Ministerio de Agricultura, Pesca y Alimentación. La gestión de dicho registro corresponde a la Secretaría General de Agricultura y Alimentación de ese mismo Ministerio, en coordinación con la Dirección General de Biodiversidad y Calidad Ambiental del Ministerio para la Transición Ecológica.
El Registro central es único en todo el territorio nacional y tiene su sede en Madrid.
El Consejo interministerial de organismos modificados genéticamente, la Comisión Nacional de Bioseguridad, los departamentos ministeriales competentes por razón de la materia y los órganos competentes de las comunidades autónomas remitirán al Registro central cuantos datos dispongan en razón de su competencia, y que resulten de la tramitación de las comunicaciones y solicitudes de autorización de utilización confinada, liberación voluntaria, y comercialización de organismos modificados genéticamente a que se refiere el Reglamento que se aprueba, antes del 31 de diciembre de cada año.
Asimismo, el registro se nutrirá de la información de la Comisión Europea y los demás Estados miembros por medio de los adecuados enlaces.
En el registro se debe reflejar la localización de los organismos modificados genéticamente objeto de liberación voluntaria con fines distintos de la comercialización, así como los que se cultiven de conformidad con lo dispuesto en la Ley 9/2003, de 25 de abril, y en el reglamento aprobado por este real decreto para su comercialización, con el fin de que los posibles efectos de dichos organismos sobre el medio ambiente puedan ser objeto de seguimiento, de acuerdo con lo dispuesto en el artículo 37.f) y en el artículo 42 del reglamento. La información relativa a la localización de cultivos de variedades vegetales modificadas genéticamente será la correspondiente a su distribución por comunidades autónomas y provincias.
Los datos estarán contenidos en un fichero apropiado para recibir, almacenar y conservar toda la información que haya de constar en el registro, y para poder recuperarla y ponerla a disposición del público.
El suministro de datos podrá realizarse por medios telemáticos, siempre que el soporte utilizado garantice la autenticidad de la comunicación y de su contenido, y quede constancia de la remisión y recepción íntegras y del momento en que se hicieron.
El acceso del público a la información recogida en el registro se efectuará teniendo en cuenta lo dispuesto en la Ley 27/2006, de 18 de julio, por la que se regulan los derechos de acceso a la información, de participación pública y de justicia en materia de medio ambiente, y de conformidad con lo previsto en las disposiciones relativas al secreto comercial e industrial, al secreto de obtención y a la protección de datos personales.
Se modifican el tercer párrafo y el último párrafo por el art. 1.1 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifica el párrafo primero por el art. 1.1 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifica el párrafo primero por el art. 1.1 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifica el párrafo primero por el art. 19.1 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 5: #dasegunda]
El funcionamiento del Consejo interministerial de organismos modificados genéticamente, de la Comisión Nacional de Bioseguridad y del Registro central regulado en la disposición adicional primera de este real decreto no supondrá incremento alguno del gasto público, y su funcionamiento será atendido con los recursos humanos y materiales de los Ministerios a que estén adscritos cada uno de ellos.
Se modifica por el art. 1.2 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
[Bloque 6: #ddunica]
Queda derogado el Real Decreto 951/1997, de 20 de junio, por el que se aprueba el Reglamento general para el desarrollo y ejecución de la Ley 15/1994, de 3 de junio, por la que se establece el régimen jurídico de utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente, a fin de prevenir los riesgos para la salud humana y el medio ambiente, y cuantas disposiciones de igual o inferior rango se opongan a lo previsto por este real decreto.
[Bloque 7: #dfprimera]
Como Punto Focal Nacional y Autoridad Nacional Competente, según el instrumento de ratificación del Protocolo de Cartagena sobre Seguridad de la Biotecnología del Convenio sobre la Diversidad Biológica, de 10 de diciembre de 2001, la Dirección General de Producciones y Mercados Agrarios de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en coordinación con la Dirección General de Biodiversidad y Calidad Ambiental del Ministerio para la Transición Ecológica, asumirá las funciones establecidas en el Reglamento (CE) n.º 1946/2003 del Parlamento Europeo y del Consejo, de 15 de julio de 2003, relativo al movimiento transfronterizo de organismos modificados genéticamente.
Se modifica por el art. 1.4 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifica por el art. 1.2 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifica por el art. 19.2 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 8: #dfsegunda]
Este real decreto y los preceptos del reglamento que se aprueba, excepto los artículos 1, 5, 6, 7, 8, 9, segundo párrafo, 24.2 y 3, 25.5 y 6, tienen carácter de legislación básica sobre protección del medio ambiente y bases de la sanidad, y se dictan al amparo del artículo 149.1 16.ª y 23.ª de la Constitución.
[Bloque 9: #dftercera]
Se faculta a las personas titulares de los Ministerios del Interior, de Industria, Comercio y Turismo, de Agricultura, Pesca y Alimentación, para la Transición Ecológica y el Reto Demográfico, de Sanidad, de Ciencia e Innovación, de Consumo y de Universidades, para dictar, en el ámbito de sus competencias, cuantas disposiciones sean necesarias para la aplicación y el desarrollo de lo establecido en este real decreto.
Se modifica por el art. 1.2 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifica por el art. 1.4 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757.
Se modifica por el art. 1.3 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifica por el art. 19.3 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 10: #dfcuarta]
El presente real decreto entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
[Bloque 11: #firma]
Dado en Madrid, a 30 de enero de 2004.
JUAN CARLOS R.
El Vicepresidente Segundo del Gobierno
y Ministro de la Presidencia,
JAVIER ARENAS BOCANEGRA
[Bloque 12: #reglamento]
[Bloque 13: #ti]
[Bloque 14: #ci]
[Bloque 15: #a1]
Este reglamento tiene por objeto dictar las normas necesarias para el desarrollo y ejecución de la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente.
[Bloque 16: #a2]
1. Este reglamento será de aplicación a las actividades de utilización confinada, liberación voluntaria con fines distintos a su comercialización y comercialización de organismos modificados genéticamente o de productos que los contengan.
2. Quedan excluidas del ámbito de aplicación de este reglamento las actividades mencionadas en el apartado anterior cuando la modificación genética de los organismos se obtenga por técnicas de mutagénesis o de fusión (incluida la de protoplastos) de células vegetales, en que los organismos resultantes puedan producirse también mediante métodos tradicionales de multiplicación o de cultivo, siempre que tales técnicas no supongan la utilización de moléculas de ácido nucleico recombinante ni de organismos modificados genéticamente.
Igualmente, quedan excluidas de este reglamento la utilización de las técnicas de fertilización «in vitro», conjugación, transducción, transformación o cualquier otro proceso natural y la inducción poliploide, siempre que no supongan la utilización de moléculas de ácido nucleico recombinante ni de organismos modificados genéticamente obtenidos mediante técnicas o métodos distintos de los que quedan excluidos en virtud del párrafo anterior.
[Bloque 17: #a3]
A los efectos de lo establecido en el artículo 2.b) de la Ley 9/2003, de 25 de abril, se consideran técnicas que dan lugar a una modificación genética las siguientes:
a) Técnicas de recombinación del ácido nucleico, que incluyan la formación de combinaciones nuevas de material genético mediante la inserción de moléculas de ácido nucleico –obtenidas por cualquier medio fuera de un organismo– en un virus, plásmido bacteriano u otro sistema de vector y su incorporación a un organismo hospedador en el que no se encuentren de forma natural pero puedan seguir reproduciéndose.
b) Técnicas que suponen la incorporación directa en un organismo de material hereditario preparado fuera del organismo, incluidas la microinyección, la macroinyección y la microencapsulación.
c) Técnicas de fusión de células (incluida la fusión de protoplastos) o de hibridación en las que se formen células vivas con combinaciones nuevas de material genético hereditario mediante la fusión de dos o más células utilizando métodos que no se producen naturalmente.
[Bloque 18: #a4]
A los efectos de lo establecido en la Ley 9/2003, de 25 de abril, y en este reglamento, además de las definiciones recogidas en el artículo 2 de aquella, se entiende por:
a) Evaluación del riesgo: la evaluación de los riesgos para la salud humana y el medio ambiente, ya sean directos o indirectos, inmediatos o diferidos, que pueden entrañar las actividades con organismos modificados genéticamente reguladas en la Ley 9/2003, de 25 de abril, y en este reglamento.
b) Producto: preparado que consista en un organismo modificado genéticamente o en una combinación de organismos modificados genéticamente, o que los contenga, y que se comercialice.
c) Trazabilidad: la capacidad de seguir el rastro de los organismos modificados genéticamente y los productos producidos a partir de organismos modificados genéticamente a lo largo de las cadenas de producción y distribución en todas las fases de su comercialización.
d) Identificador único: código numérico o alfanumérico sencillo cuyo objeto es identificar cada organismo modificado genéticamente conforme a la transformación genética autorizada de la que procede, y facilitar que se recabe información específica de aquéllos.
e) Operador: toda persona física o jurídica que comercialice un producto o reciba un producto comercializado en la Unión Europea, tanto de un Estado miembro como de un tercer país, en cualquier fase de su producción o distribución, exceptuando el consumidor final.
f) Consumidor final: el último consumidor que no vaya a utilizar el producto como parte de una operación comercial.
g) Producto preenvasado: condición de un artículo unitario para la venta, integrado por un producto y el envase en que haya sido colocado antes de ponerlo en venta y que lo cubra de forma total o parcial, siempre que el contenido no pueda modificarse sin abrir o alterar el envase.
h) Técnicas de autoclonación: la extracción de secuencias de ácido nucleico de una célula de un organismo, que puede ir o no seguida de la reinserción total o parcial de dicho ácido nucleico (o de un equivalente sintético), con o sin fases enzimáticas o mecánicas previas, en células de la misma especie o de una especie que presente características filogenéticas muy similares, que puedan intercambiar material genético por procesos fisiológicos naturales, siempre que sea improbable que el organismo resultante sea patógeno para las personas, los animales o los vegetales. La autoclonación puede incluir el empleo de vectores recombinantes en relación con los cuales se disponga de una larga historia de utilización segura en los organismos correspondientes.
[Bloque 19: #cii]
[Bloque 20: #a5]
1. Las competencias que atribuyen a la Administración General del Estado en materia de autorizaciones e informe los apartados 1 a 3 del artículo 3 y la disposición adicional segunda de la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente, serán ejercidas por el Consejo interministerial de organismos modificados genéticamente, adscrito al Ministerio de Agricultura, Pesca y Alimentación, y por la Comisión Nacional de Bioseguridad, adscrita al Ministerio para la Transición Ecológica.
2. El Consejo interministerial de organismos modificados genéticamente y la Comisión Nacional de Bioseguridad ajustarán sus actuaciones y funcionamiento a lo dispuesto en la sección 3.ª del capítulo II del título preliminar de la Ley 40/2015, de 1 de octubre, de Régimen Jurídico del Sector Público.
3. Corresponde a las comunidades autónomas el ejercicio de las funciones reguladas en el artículo 4 de la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente.
Así mismo, de conformidad con la disposición final segunda de la ley anteriormente citada, corresponde a las comunidades autónomas la función de formular observaciones respecto de las solicitudes de autorización de liberaciones voluntarias y de comercialización cuya resolución ha de otorgar el Consejo interministerial de organismos modificados genéticamente.
4. Las administraciones públicas promoverán que se habiliten los medios necesarios para hacer efectiva la tramitación electrónica de las obligaciones de comunicación, información y de los procedimientos administrativos que deriven de esta norma.
Se modifican los apartados 1 y 2 por el art. 2.1 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se añade el apartado 4 por el art. 19.4 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 21: #s1]
[Bloque 22: #a6]
1. El Consejo Interministerial de organismos modificados genéticamente estará integrado por los siguientes miembros:
a) Presidencia: la persona titular de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación.
b) Vicepresidencia: la persona titular de la Dirección General Producciones y Mercados Agrarios del Ministerio de Agricultura, Pesca y Alimentación.
c) Vocales:
1.º Dos representantes del Ministerio de Agricultura, Pesca y Alimentación, uno de la Dirección General de la Industria Alimentaria y otro de la Dirección General de Sanidad de la Producción Agraria.
2.º Un representante del Ministerio del Interior, de la Dirección General de Protección Civil y Emergencias.
3.º Un representante del Ministerio de Industria, Comercio y Turismo, de la Dirección General de Política Comercial.
4.º Dos representantes del Ministerio de Ciencia e Innovación uno en representación de la Agencia Estatal de Investigación y otro de la Secretaría General de Investigación.
5.º Un representante del Ministerio de Universidades, de la Secretaría General de Universidades.
6.º Dos representantes del Ministerio de Sanidad, uno en representación de la Dirección General de Salud Pública y otro de la Agencia Española de Medicamentos y Productos Sanitarios.
7.º Un representante de la Agencia Española de Seguridad Alimentaria y Nutrición, en representación del Ministerio de Consumo.
8.º Un representante del Ministerio para la Transición Ecológica y el Reto Demográfico.
9.º Formará igualmente parte del Consejo, con voz, pero sin voto, quien ostente la Presidencia de la Comisión Nacional de Bioseguridad.
d) Actuará como Secretario, con voz, pero sin voto, un funcionario de carrera de la Dirección General de Producciones y Mercados Agrarios de, al menos, nivel 28.
2. Las personas titulares de las vocalías tendrán, al menos, rango de Director General o asimilado, salvo en el caso del representante de la Agencia Española de Seguridad Alimentaria y Nutrición, en que podrá ser la persona titular de su Dirección Ejecutiva con independencia de su rango. Serán propuestas por las personas titulares de los Ministerios correspondientes, y nombrados por la persona titular del Ministerio de Agricultura, Pesca y Alimentación.
Con sujeción al procedimiento establecido en el párrafo anterior, se podrán designar suplentes en las vocalías, en los supuestos de vacante, ausencia o enfermedad de las personas titulares, siempre que recaiga en una persona designada entre el personal funcionario de carrera destinado en la unidad correspondiente, que pertenezca a un Cuerpo o Escala clasificado en el subgrupo A1.
3. Sin perjuicio de lo dispuesto en los apartados anteriores, cuando la naturaleza o la importancia de los asuntos a tratar así lo requiera, podrán asistir a las reuniones del Consejo Interministerial de Organismos Modificados Genéticamente, con voz, pero sin voto, los titulares de otros órganos directivos cuyo ámbito de gestión tenga relación con la materia a tratar.
4. El Consejo se reunirá cuantas veces sea necesario para el cumplimiento de sus funciones. De conformidad con lo dispuesto en el artículo 17 de la Ley 40/2015, de 1 de octubre, se podrán convocar, celebrar sesiones, adoptar acuerdos y remitir actas, tanto de forma presencial como a distancia. Para la adopción de acuerdos a distancia, se remitirá la petición de informe, el orden del día a tratar y la documentación correspondiente a todos los miembros del Consejo, por vía electrónica y con un plazo de, al menos, ocho días hábiles de antelación, que, por motivos de urgencia, podrá ser rebajado hasta cinco.
En las actas que se levantaren para constancia de estas reuniones, se incorporarán las comunicaciones que hayan tenido lugar, tanto para la convocatoria como para las deliberaciones y la adopción de decisiones. Los miembros del consejo podrán hacer constar en acta su voto particular en relación a los acuerdos alcanzados.
El procedimiento a distancia será el ordinario, sin perjuicio de las reuniones presenciales que estime preciso convocar la persona titular de la Presidencia.
5. En el marco del Consejo, se crea un Comité de Participación, adscrito a aquel, en el que se encontrarán representados los sectores interesados, las organizaciones profesionales agrarias de ámbito nacional, las cooperativas agroalimentarias, las organizaciones de consumidores y usuarios, las organizaciones conservacionistas, en total, hasta un máximo de quince miembros, designados todos ellos por el Ministerio de Agricultura, Pesca y Alimentación, a propuesta de las entidades respectivas. Su composición y funcionamiento se regulan en la Orden ARM/2616/2010, de 5 de octubre.
El Comité de Participación será el órgano de interlocución e información entre los ciudadanos y la Administración General del Estado, en materia de organismos modificados genéticamente. Los miembros del comité serán debidamente informados de los acuerdos e informes adoptados al objeto de permitirles emitir el juicio que en cada caso resulte procedente.
Sus funciones serán las de asesorar al Consejo Interministerial en cuantas cuestiones le sean solicitadas por éste, elevar a la consideración del mismo cuantas cuestiones se estimen oportunas, asegurar la participación e información pública en las actuación del Consejo así como permitir a éste recabar y tener conocimiento de la posición de los diferentes sectores implicados. En los casos en que se vayan a tratar en el comité de participación asuntos que afecten a un sector específico, se podrá solicitar la asistencia de las principales asociaciones u organizaciones nacionales representativas del respectivo sector, así como de aquellos expertos de reconocido prestigio que, en razón de la materia, resulte justificado.
Se modifican los apartados 1, 2 y 4 por el art. 2.1 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifican los apartados 1, 2 y 5 por el art. 2.2 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifica por el art. 2.1 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifica por el art. 19.5 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
Redactados los apartados 1.c) y d) conforme a la corrección de errores publicada en BOE núm. 283, de 23 de noviembre de 2010. Ref. BOE-A-2010-17977
[Bloque 23: #a7]
1. Corresponde al Consejo Interministerial de Organismos Modificados Genéticamente el otorgamiento de las autorizaciones a que se refiere el artículo 3 de la Ley 9/2003, de 25 de abril, así como las demás funciones que se le asignan en este reglamento.
2. Cuando las autorizaciones de utilización confinada y liberación voluntaria con fines distintos a la comercialización tengan por objeto alguna de las actividades a que se refiere el artículo 3.2.a) de la ley anteriormente citada, el otorgamiento de la autorización estará condicionado a la conformidad de la representación del Ministerio de Sanidad, salvo en el caso de los medicamentos veterinarios, cuya autorización requerirá la conformidad de los representantes de los Ministerios de Sanidad y de Agricultura, Pesca y Alimentación.
3. Si las autorizaciones tuvieran por objeto la realización de actuaciones de utilización confinada y liberación voluntaria de organismos modificados genéticamente (OMG), en los supuestos que deriven de las actividades de investigación científica y técnica contempladas en el artículo 42 de la Ley 14/2011, de 1 de junio, de la Ciencia, la Tecnología y la Innovación, y en el marco de programas de investigación realizados por órganos u organismos dependientes de la Administración General del Estado, el otorgamiento de la autorización queda supeditado a la conformidad de la representación del Ministerio de Ciencia e Innovación y del Ministerio de Universidades.
4. Para las autorizaciones relacionadas con el examen técnico para la inscripción de variedades comerciales a que se refiere el artículo 3.2.c) de la Ley 9/2003, de 25 de abril, será precisa la conformidad de la representación de la Dirección General de Producciones y Mercados Agrarios del Ministerio de Agricultura, Pesca y Alimentación.
5. Las resoluciones del Consejo Interministerial de Organismos Modificados Genéticamente que otorguen o denieguen las autorizaciones pondrán fin a la vía administrativa.
6. Asimismo, le corresponden al Consejo Interministerial las siguientes funciones:
a) Decidir el contenido de los documentos a remitir a los organismos internacionales, y establecer la posición española ante los mismos, incluyendo la posición ante las instancias de la Unión Europea.
b) Realizar un informe anual sobre las actividades llevadas a cabo en materia de organismos modificados genéticamente con base en la información facilitada por los órganos de la Administración General del Estado competentes en esta materia, las comunidades autónomas y las empresas afectadas. El citado informe será público una vez aprobado.
Se modifican los apartados 2 y 3 por el art. 2.2 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifican los apartados 2 a 4 por el art. 2.3 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifican los apartados 3 y 4 por el art. 2.2 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifica por el art. 19.5 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 24: #s2]
[Bloque 25: #a8]
1. La Comisión Nacional de Bioseguridad, prevista en la disposición adicional segunda de la Ley 9/2003, de 25 de abril, es un órgano colegiado de carácter consultivo de la Administración General del Estado y de las comunidades autónomas.
2. La Comisión Nacional de Bioseguridad estará compuesta por los siguientes miembros:
a) Presidente: la persona titular de la Dirección General de Calidad y Evaluación Ambiental del Ministerio para la Transición Ecológica y el Reto Demográfico.
b) Vicepresidente: designado de entre las personas titulares de las vocalías en representación de la Administración General del Estado por acuerdo del Pleno de la Comisión, que será nombrado por la persona titular del Ministerio para la Transición Ecológica y el Reto Demográfico.
c) Vocales:
1.º En representación de la Administración General del Estado:
Una persona designada entre el personal funcionario de carrera representante de la Dirección General de Protección Civil y Emergencias del Ministerio del Interior.
Una persona designada entre el personal funcionario de carrera representante de la Secretaría General de Universidades del Ministerio de Universidades.
Cuatro personas designadas entre el personal funcionario de carrera representantes de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, expertos en tecnología alimentaria, agricultura, ganadería, sanidad vegetal y animal.
Dos personas designadas entre el personal funcionario de carrera de la Secretaría de Estado de Medio Ambiente del Ministerio para la Transición Ecológica y el Reto Demográfico, expertos en biodiversidad y bioseguridad.
Tres personas designadas entre el personal funcionario de carrera en representación del Ministerio de Sanidad, expertos en seguridad alimentaria, medicamentos de uso humano, medicamentos veterinarios y salud pública.
Dos personas designadas entre el personal funcionario de carrera en representación del Ministerio de Consumo, expertos en seguridad alimentaria y técnicas analíticas, uno de ellos de la Agencia Española de Seguridad Alimentaria y Nutrición.
Cuatro personas designadas entre el personal funcionario de carrera en representación del Ministerio de Ciencia e Innovación, expertos en investigación científica y técnica, proyectos y actividades de investigación en biotecnología, biomedicina y en tecnologías agroalimentarias.
Una persona designada entre el personal funcionario de carrera del Ministerio de Industria, Comercio y Turismo, experto en comercio exterior.
Una persona designada, entre el personal funcionario de carrera, en representación del Instituto Nacional de Seguridad y Salud en el Trabajo, del Ministerio de Trabajo y Economía Social.
Las personas titulares de las vocalías serán designados por las personas titulares de los órganos de los que dependen jerárquicamente. La Comisión Nacional de Bioseguridad comunicará dicha designación al Consejo Interministerial de OMG.
2.º Una vocalía por cada una de las comunidades autónomas, previa comunicación al Consejo Interministerial de OMG.
d) Actuará como Secretario, con voz, pero sin voto, una persona designada entre el personal funcionario de carrera de la Dirección General de Calidad y Evaluación Ambiental de, al menos, nivel 28.
3. La Comisión Nacional de Bioseguridad podrá incorporar como miembros permanentes hasta un máximo de diez expertos de instituciones científicas, en las materias comprendidas en la Ley 9/2003, de 25 de abril. El nombramiento de los mismos deberá ser objeto de informe favorable por parte del Consejo Interministerial de OMG, a propuesta de la Presidencia de la Comisión Nacional de Bioseguridad.
4. Igualmente, la Comisión, a propuesta de su Presidencia, podrá invitar a participar en sus reuniones, con voz pero sin voto, a aquellos científicos o expertos cuya asistencia quede justificada para determinados temas concretos.
Se modifica el apartado 2 por el art. 2.3 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifican los apartados 2 y 3 por el art. 2.4 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifica el apartado 2 por el art. 2.3 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifica por el art. 19.5 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 26: #a9]
1. La Comisión Nacional de Bioseguridad informará preceptivamente las solicitudes de autorización que corresponde otorgar a la Administración General del Estado y a las comunidades autónomas.
2. Además, ejercerá las siguientes funciones:
a) Informar sobre la clasificación del tipo más adecuado para la utilización confinada de organismos modificados genéticamente propuesta, en el supuesto a que se refiere el artículo 12.3.
b) Informar sobre si los datos y documentos aportados son completos y exactos, si las medidas relativas a la gestión de residuos, seguridad y respuesta en caso de emergencia son las adecuadas y si la actividad cuya realización se pretende se ajusta a las disposiciones de la Ley 9/2003, de 25 de abril, y de este reglamento, tal y como se establece en el artículo 16.1 de este último.
c) Informar sobre si los proyectos de utilización confinada de organismos modificados genéticamente han de someterse a información pública, tal y como se establece en el artículo 16.2.d.
d) Informar con carácter previo a la adopción de la resolución correspondiente por los órganos competentes, en los supuestos previstos en los artículos 15, 17.3, 19.4, 24, 25.5 y 6, 33.2, 35.1, 40.2, 42.1, 44.2, 46.2 y 50.
e) Informar sobre las propuestas, el desarrollo y aplicación de los planes de seguimiento en los supuestos a que se refieren los artículos 32.2, 37 y 42.
f) Informar sobre las demás cuestiones que se sometan a su consideración por el Consejo interministerial de organismos modificados genéticamente o por el órgano competente de las comunidades autónomas.
Se modifica por el art. 19.5 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 27: #a10]
1. La Comisión Nacional de Bioseguridad podrá actuar en Pleno o mediante grupos de trabajo, y se reunirá tantas veces como sea preciso para informar las solicitudes de autorización para actividades realizadas con organismos modificados genéticamente y para el desempeño de las demás funciones a que se refiere el artículo anterior.
2. Bajo la dependencia de la Comisión se podrán crear cuantos grupos de expertos se estimen necesarios para la realización de las actividades de apoyo científico y técnico precisas para el cumplimiento de sus funciones.
El acuerdo de creación de dichos grupos, que no podrán tener carácter permanente, se efectuará por el Pleno de la Comisión y establecerá su composición, objetivos y plazos de actuación, correspondiendo la coordinación de los grupos que se creen al presidente de la Comisión.
3. Los miembros de la Comisión Nacional de Bioseguridad y de los grupos de expertos canalizarán toda relación institucional derivada de su pertenencia a ésta a través de su presidente, y deberán abstenerse de llevar a cabo actividades de comunicación de riesgos, así como cualquier tipo de manifestaciones o declaraciones en relación con sus actividades de informe, estudio o evaluación, sin perjuicio del deber de información de los representantes de las Administraciones públicas a sus departamentos.
4. La Comisión Nacional de Bioseguridad informará al Consejo interministerial de organismos modificados genéticamente sobre sus actuaciones, los informes elaborados y las actas de sus sesiones.
Se modifica por el art. 19.5 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 28: #tii]
[Bloque 29: #ci-2]
[Bloque 30: #a11]
1. Se entiende por utilización confinada cualquier actividad por la que se modifique el material genético de un organismo o por la que éste, así modificado, se cultive, almacene, emplee, transporte, destruya o elimine, siempre que en la realización de tales actividades se utilicen medidas de confinamiento, con el fin de limitar su contacto con la población y el medio ambiente.
2. Quedan excluidas de las obligaciones establecidas en este capítulo:
a) Las modificaciones genéticas obtenidas por técnicas de autoclonación y de fusión celular, incluida la de protoplastos, tanto de especies procarióticas con intercambio de material genético por procesos fisiológicos conocidos, como de células de cualquier especie eucariótica, incluida la producción de hibridomas, siempre que tales técnicas o métodos no supongan la utilización de moléculas de ácido nucleico recombinante ni de organismos modificados genéticamente obtenidos mediante técnicas o métodos distintos de los que quedan excluidos en virtud del artículo 2.2, párrafo primero.
b) Las utilizaciones confinadas que incluyan únicamente tipos de organismos modificados genéticamente de naturaleza inocua determinada de conformidad con la Decisión 2001/204/CE del Consejo, de 8 de marzo de 2001, y demás disposiciones comunitarias que la modifiquen o complementen.
3. Lo dispuesto en este capítulo no será tampoco de aplicación al almacenamiento, cultivo, transporte, destrucción, eliminación ni utilización de organismos modificados genéticamente que ya se hayan comercializado con arreglo al capítulo III de este título o a otra norma en la que se exija una evaluación del riesgo para la salud humana y el medio ambiente equivalente a la establecida en este capítulo, siempre que la utilización confinada se ajuste, en caso de haberlas, a las condiciones de la autorización de puesta en el mercado.
[Bloque 31: #a12]
1. Las actividades de utilización confinada de organismos modificados genéticamente se clasificarán en función de la evaluación previa de los riesgos para la salud humana y el medio ambiente en los siguientes tipos:
a) Tipo 1. Actividades de riesgo nulo o insignificante: aquellas en las cuales el grado 1 de confinamiento es suficiente para proteger la salud humana y el medio ambiente.
b) Tipo 2. Actividades de bajo riesgo: aquellas en las cuales el grado 2 de confinamiento es suficiente para proteger la salud humana y el medio ambiente.
c) Tipo 3. Actividades de riesgo moderado: aquellas en las cuales el grado 3 de confinamiento es suficiente para proteger la salud humana y el medio ambiente.
d) Tipo 4. Actividades de alto riesgo: aquellas en las cuales el grado 4 de confinamiento es suficiente para proteger la salud humana y el medio ambiente.
2. La evaluación del riesgo para la salud humana y el medio ambiente se llevará a cabo de conformidad con lo establecido en el anexo I.
3. Cuando existan dudas razonables sobre la clasificación de una determinada actividad, se aplicarán las medidas correspondientes al tipo de riesgo más elevado. No obstante, el órgano competente podrá autorizar que la actividad se realice aplicando medidas menos rigurosas, siempre que el responsable de la operación lo haya comunicado previamente, aportando pruebas que lo justifique.
4. A efectos de lo establecido en el apartado anterior, cuando la competencia corresponda a la Administración General del Estado, la comunicación se dirigirá al Director General de Desarrollo Sostenible del Medio Rural, en su calidad de presidente del Consejo interministerial de organismos modificados genéticamente.
El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de la Comisión Nacional de Bioseguridad dicha comunicación. El Consejo interministerial deberá resolver sobre la autorización solicitada, previo informe de la Comisión, en el plazo de un mes.
Se modifica el apartado 4 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 32: #a13]
1. Toda persona física o jurídica que pretenda realizar una actividad de utilización confinada de organismos modificados genéticamente estará obligada a:
a) Realizar una evaluación previa del riesgo para la salud humana y el medio ambiente, conforme a lo establecido en el artículo anterior.
b) Llevar un registro de la evaluación del riesgo para la salud humana y el medio ambiente que deberá presentarse al órgano competente como parte de las comunicaciones contempladas en el artículo 14, así como cuando éste lo solicite. El citado registro contendrá, como mínimo, la siguiente información:
1.º Fecha de presentación de la comunicación a que se refiere el artículo 14.1.
2.º Nombre del titular de la actividad y de las personas responsables de la supervisión y de la seguridad.
3.º Descripción de la actividad: objetivo y duración de ésta.
4.º Identificación de las características del organismo modificado genéticamente que pueden causar efectos adversos en la salud humana o el medio ambiente.
5.º Clasificación final de la actividad.
6.º Fechas de las revisiones periódicas de las instalaciones a que se refiere el párrafo f) de este apartado que realice el titular de la actividad.
c) Cumplir las normas específicas de seguridad e higiene profesional y aplicar los principios de las buenas prácticas de microbiología, conforme a lo establecido en el anexo II.
d) Aplicar los principios generales y las medidas de confinamiento y de protección que correspondan al tipo de utilización confinada, previstas en el anexo II, de manera que la exposición del lugar de trabajo y del medio ambiente a los organismos modificados genéticamente sea la menor posible y se garantice un alto grado de seguridad.
e) Elaborar los planes de emergencia y de vigilancia de las instalaciones, en los supuestos previstos en el artículo 20.
f) Revisar periódicamente las medidas de confinamiento y de protección aplicadas, así como la evaluación del riesgo realizada. Dicha revisión deberá realizarse siempre que las medidas de confinamiento aplicadas ya no resulten adecuadas o el tipo asignado a las utilizaciones confinadas ya no sea correcto, o cuando haya motivos para suponer que, a la luz de nuevos conocimientos científicos o técnicos, la evaluación del riesgo ya no es adecuada.
2. El transporte por cualquier medio de organismos modificados genéticamente sólo requerirá que se realice una evaluación del riesgo para la salud humana y el medio ambiente y que se cumplan las normas específicas de seguridad e higiene profesional.
[Bloque 33: #a14]
1. Las personas físicas o jurídicas que se propongan utilizar por primera vez instalaciones específicas para utilizaciones confinadas de organismos modificados genéticamente estarán obligadas a comunicarlo previamente al órgano competente, sin perjuicio de las demás autorizaciones o licencias que sean exigibles de acuerdo con la legislación vigente.
Dicha comunicación deberá contener la información establecida en la parte A del anexo III.
2. Para las primeras o sucesivas utilizaciones confinadas del tipo 2, tras la comunicación a la que hace referencia el apartado 1, deberá presentarse una comunicación que contendrá la información establecida en la parte B del anexo III.
3. Para las primeras o sucesivas utilizaciones confinadas de los tipos 3 y 4, tras la comunicación a la que hace referencia el apartado 1, deberá presentarse una comunicación que contendrá la información establecida en la parte C del anexo III.
[Bloque 34: #a15]
1. Cuando la competencia corresponda a la Administración General del Estado, las comunicaciones reguladas en este capítulo se dirigirán al Director General de Desarrollo Sostenible del Medio Rural, en su calidad de presidente del Consejo interministerial de organismos modificados genéticamente.
Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de los de la Comisión Nacional de Bioseguridad dicha comunicación.
2. Cuando la competencia corresponda a las comunidades autónomas, el titular de la actividad deberá presentar ante el órgano competente de ésta la correspondiente comunicación.
El órgano competente, una vez realizadas las actuaciones de comprobación documental oportunas, remitirá inmediatamente copia de la citada comunicación al Director General de Desarrollo Sostenible del Medio Rural quien, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de la Comisión Nacional de Bioseguridad copia de la comunicación para que ésta emita el preceptivo informe de conformidad con lo establecido en el artículo 9.
Se modifica por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 35: #a16]
1. El órgano competente comprobará que la información, los datos y documentos aportados son completos y exactos, que la evaluación del riesgo para la salud humana y el medio ambiente y el tipo de utilización confinada de organismos modificados genéticamente son correctos y que, en su caso, las medidas relativas al confinamiento, las demás medidas de protección, la gestión de los residuos, los planes de emergencia y vigilancia son los adecuados, y que todo ello, así como la actividad que se pretende realizar, se ajusta a las disposiciones de la Ley 9/2003, de 25 de abril, y de este reglamento.
2. En su caso, el órgano competente podrá:
a) Exigir a los responsables de la utilización confinada de organismos modificados genéticamente que proporcionen información adicional, que modifiquen las condiciones o el tipo asignado a la utilización confinada propuesta. En este caso, el órgano competente podrá exigir que la utilización confinada propuesta no se inicie y, si ya se ha iniciado, se suspenda o se le ponga fin hasta que el órgano competente haya dado su autorización con arreglo a la información complementaria obtenida o a la modificación de las condiciones para la utilización confinada.
b) Limitar el período en que se permite la utilización confinada, o supeditarla a determinadas condiciones específicas.
c) Consultar a los expertos, instituciones o Administraciones públicas cuando por las características o naturaleza de la actividad que se vaya a desarrollar se estime conveniente.
d) Someter la comunicación a información pública, siendo ésta preceptiva si se trata de operaciones de los tipos 3 y 4. El plazo ordinario de información pública será de treinta días, que podrá reducirse hasta un mínimo de veinte días por resolución del órgano competente, por motivos de necesidad. En todo caso se considera necesidad cuando la actividad esté relacionada con la prevención o contención de agentes infecciosos, procesos o situaciones que puedan tener repercusiones para la salud y para los que se precise actuaciones inmediatas o a corto plazo. Todo ello sin perjuicio de la posible aplicación del procedimiento de la tramitación de urgencia, en los términos del artículo 33 de la Ley 39/2015, de 1 de octubre.
Se modifica la letra d) del apartado 2 por el art. 2.4 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
[Bloque 36: #a17]
1. Las actividades de utilización confinada de tipo 1 podrán ser realizadas por los titulares de las actividades:
a) Inmediatamente después de la presentación de la comunicación de primera utilización de una instalación para actividades de utilización confinada de tipo 1 a que hace referencia el artículo 14.1.
b) Si la instalación ya ha sido objeto de una comunicación conforme al artículo 14.1, las actividades de utilización confinada sucesivas del tipo 1 no requerirán ninguna otra comunicación.
Las personas físicas y jurídicas que se propongan realizar estas operaciones estarán obligadas a llevar un registro de las evaluaciones del riesgo para la salud humana y el medio ambiente de dichas operaciones, que deberán facilitar al órgano competente cuando éste lo solicite. El registro contendrá como mínimo la información indicada en el artículo 13.1.b).
2. Las utilizaciones confinadas del tipo 2 podrán ser realizadas por los titulares de las actividades:
a) Si las instalaciones no han sido objeto de una comunicación previa relativa a actividades de utilización confinada de los tipos 2 o siguientes, la utilización confinada del tipo 2 podrá iniciarse, salvo indicación en contrario de la autoridad competente, 45 días después de la presentación de la comunicación regulada en el artículo 14.2, o antes si el órgano competente así lo autoriza.
b) Si las instalaciones han sido objeto de una comunicación previa relativa a actividades de utilización confinada de los tipos 2 o siguientes y se cumplen los requisitos de la autorización, la utilización confinada del tipo 2 podrá iniciarse inmediatamente después de la nueva comunicación.
3. A efectos de lo establecido en los apartados anteriores y de conformidad con lo establecido en el artículo 9, el órgano competente adoptará la correspondiente resolución, previo informe de la Comisión Nacional de Bioseguridad.
[Bloque 37: #a18]
1. Requerirán autorización expresa del órgano competente las actividades de utilización confinada de los tipos 3 y 4.
2. Las actividades de utilización confinada de tipo 2 estarán también sujetas a autorización expresa en los siguientes supuestos:
a) Cuando el órgano competente solicite al titular mayor información que la aportada en su comunicación, o cuando se modifiquen las condiciones o el tipo asignado a la utilización confinada propuesta.
b) Cuando así lo solicite el interesado en realizar una utilización confinada de tipo 2 en instalaciones que hayan sido objeto de alguna de las comunicaciones a que se refieren los apartados 2 y 3 del artículo 14.
[Bloque 38: #a19]
1. El órgano competente deberá emitir una resolución sobre la autorización de las actividades de utilización confinada previstas en el artículo 18.2 en el plazo de 45 días a partir de la comunicación a que hace referencia el artículo 14.2.
2. El órgano competente deberá emitir una resolución sobre la autorización de las actividades de utilización confinada de los tipos 3 y 4, previstas en el artículo 18.1:
a) En el plazo de 45 días después de la presentación de la comunicación regulada en el artículo 14.3, si las instalaciones han sido previamente autorizadas para ejecutar utilizaciones confinadas de los tipos 3 y 4 y se cumplen los requisitos de la autorización para el mismo tipo de utilización confinada o un tipo superior al de la utilización confinada que se pretende realizar.
b) En el plazo de 90 días después de la presentación de la comunicación regulada en el artículo 14.3, si las instalaciones no han sido objeto de una autorización previa para ejecutar utilizaciones confinadas de los tipos 3 y 4.
3. No obstante, el órgano competente podrá autorizar expresamente la realización de las actividades antes de los plazos señalados en los apartados anteriores.
4. A efectos de lo establecido en este artículo, y de conformidad con lo regulado en el artículo 9, el órgano competente adoptará la correspondiente resolución, previo informe de la Comisión Nacional de Bioseguridad.
5. Las resoluciones deberán notificarse dentro del plazo para la adopción de las autorizaciones reguladas en este artículo.
[Bloque 39: #a20]
1. Cuando sea necesario, a juicio del órgano competente, y en todo caso antes de que comience una operación de utilización confinada de organismos modificados genéticamente de los tipos 3 y 4, y en cualquier caso cuando un fallo en las medidas de confinamiento pudiera ocasionar un peligro grave para la salud humana y el medio ambiente, se deberá elaborar un plan de emergencia sanitaria y de vigilancia epidemiológica y medioambiental, salvo en los casos en que se haya elaborado un plan de emergencia de estas características en virtud de la legislación sectorial vigente aplicable a la instalación. Estos planes incluirán las actuaciones que se hayan de seguir para la protección de la salud humana y del medio ambiente en el caso de que se produzca un accidente en el exterior de las instalaciones donde radique la actividad.
2. Estos planes de emergencia sanitaria y de vigilancia epidemiológica y medioambiental serán elaborados por el órgano que designe la comunidad autónoma donde radiquen las instalaciones, y se remitirán al órgano competente. Los planes se elaborarán teniendo en cuenta los datos suministrados por los titulares de las actividades de utilización confinada, de acuerdo con lo establecido en el anexo III, y tendrán, al menos el siguiente contenido:
a) Análisis de los riesgos que distintas hipótesis de accidente pueden suponer para la salud humana y el medio ambiente en el exterior de las instalaciones.
b) Actuaciones de tipo sanitario, epidemiológico y medioambiental que se deban seguir, en caso de accidente.
c) Organización de los servicios, medios y recursos necesarios para el desempeño de las actuaciones previstas.
d) Identificación de la persona u órgano al que deba facilitar la información sobre las medidas de seguridad y el comportamiento que deban observar los posibles afectados, en caso de accidente, en el exterior de las instalaciones, de conformidad con el apartado 5 de este artículo.
e) Identificación de la persona u órgano al que el titular de la actividad deberá comunicar, en caso de accidente, la información señalada en el artículo 21.
f) Identificación de la persona u órgano al que se atribuyen las funciones de dirección y coordinación de las actuaciones que se deban seguir en caso de accidente, de acuerdo con lo establecido en el artículo 21.
3. Cuando el Consejo interministerial de organismos modificados genéticamente, en los supuestos del artículo 3.2 de la Ley 9/2003, de 25 de abril, estime que se debe elaborar un plan de emergencia sanitaria y de vigilancia epidemiológica y medioambiental, enviará al órgano designado por la comunidad autónoma correspondiente la información suministrada por los titulares de las actividades de utilización confinada, de acuerdo con lo establecido en el anexo III, para que proceda a la elaboración de dicho plan.
4. Los titulares de las actividades elaborarán planes de emergencia y vigilancia interior que contemplen las adecuadas medidas de prevención de riesgos y las actuaciones ante situaciones de emergencia, de alarma o de socorro, y la evacuación en el interior de las instalaciones.
5. Cuando así venga exigido por la normativa aplicable en el ámbito de la protección civil, el órgano al que corresponda la aprobación de los planes de emergencia, previamente a que recaiga ésta, dará traslado de dichos planes al órgano competente en materia de protección civil, a efectos de recabar su aprobación o de establecer la coordinación de actuaciones y recursos según proceda.
6. El órgano competente se asegurará de que, antes de iniciarse una utilización confinada, las personas, organismos y autoridades que puedan verse afectados en caso de accidente estén adecuadamente informados, sin que deban solicitarlo, sobre los planes de emergencia y vigilancia previstos en los apartados anteriores. Esta información se repetirá y actualizará con la periodicidad adecuada. En cualquier caso, tal información será pública y de libre acceso para el ciudadano.
7. La información sobre las medidas de seguridad y el comportamiento que, en caso de accidente, se deberán observar en el interior de la instalación será proporcionada por los titulares de las actividades.
La información sobre las medidas de seguridad y el comportamiento que se deban observar en el exterior de la instalación se facilitará por la persona u órgano que, en cada caso, se establezca en el correspondiente plan de emergencia sanitaria y de vigilancia epidemiológica y medioambiental.
8. La información indicada en el apartado 2 se comunicará a la Dirección General de Desarrollo Sostenible del Medio Rural para que ésta informe a los demás Estados miembros de la Unión Europea interesados acerca de las medidas de seguridad adoptadas de conformidad con este artículo. Asimismo, la Dirección General de Desarrollo Sostenible del Medio Rural efectuará consultas sobre la aplicación de planes de emergencia sanitaria y de vigilancia epidemiológica a los Estados miembros que puedan verse afectados en caso de accidente.
Se modifica el apartado 8 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 40: #a21]
1. En caso de accidente, el titular de la actividad estará obligado a comunicarlo inmediatamente al órgano competente, así como a la persona u órgano que a estos efectos se haya establecido en el plan de emergencia sanitaria y de vigilancia epidemiológica y medioambiental de la comunidad autónoma, facilitando la siguiente información:
a) Las circunstancias del accidente.
b) La identidad y cantidad de los organismos modificados genéticamente de que se trate.
c) Cualquier información necesaria para evaluar los efectos del accidente sobre la salud de la población y sobre el medio ambiente.
d) Las medidas de emergencia que se hayan adoptado tras el accidente.
A efectos de este apartado, cuando la competencia para recibir la comunicación corresponda a la Administración General del Estado, la comunicación se dirigirá a la persona titular de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en su calidad de Presidente del Consejo Interministerial de OMG, y podrá presentarse por cualquiera de los medios previstos en el artículo 16.4 de la Ley 39/2015, de 1 de octubre, del Procedimiento Administrativo Común de las Administraciones Públicas.
El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de los de la Comisión Nacional de Bioseguridad dicha comunicación.
2. Cuando, conforme a lo establecido en el apartado anterior, se informe de un accidente, la persona u órgano que tenga atribuidas en el plan de emergencia sanitaria y de vigilancia epidemiológica y medioambiental las funciones de dirección y coordinación de las actuaciones que deban seguirse en caso de accidente, dispondrá su aplicación y la movilización de los servicios, medios y recursos previstos en aquél que resulten necesarios para:
a) Adoptar todas las medidas de emergencia necesarias para la protección de la salud de las personas y del medio ambiente.
b) Recopilar la información necesaria para realizar un análisis completo del accidente y, cuando proceda, formular recomendaciones para evitar que se produzcan accidentes similares en el futuro y limitar sus consecuencias.
3. A efectos de su comunicación a la Comisión Europea, las personas u órgano que tengan atribuidas las funciones de dirección y coordinación de los planes de emergencia sanitaria y de vigilancia epidemiológica y medioambiental remitirán a la Dirección General de Desarrollo Sostenible del Medio Rural la información que hayan recibido, de acuerdo con lo establecido en el apartado 1, y proporcionarán datos detallados acerca de las circunstancias del accidente, de la identidad y la cantidad de los organismos modificados genéticamente que se hubieran liberado, de las medidas de emergencia aplicadas y de su eficacia, así como un análisis del accidente con recomendaciones para limitar sus efectos y evitar accidentes similares en el futuro.
La Dirección General de Desarrollo Sostenible del Medio Rural informará lo antes posible a la Comisión Europea e inmediatamente a los Estados miembros de la información señalada en el párrafo anterior.
4. Sin perjuicio de lo dispuesto en los apartados anteriores, los titulares de la actividad deberán comunicar todo accidente al órgano competente en materia de protección civil, aportando la información y adoptando las medidas de emergencia que fueran exigibles, conforme a lo dispuesto en la normativa reguladora de esta materia.
Se modifica el apartado 1 por el art. 2.5 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifican los apartados 1 y 3 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 41: #cii-2]
[Bloque 42: #a22]
1. Se entiende por liberación voluntaria la introducción deliberada en el medio ambiente de un organismo o combinación de organismos modificados genéticamente sin que hayan sido adoptadas medidas específicas de confinamiento, para limitar su contacto con la población y el medio ambiente y proporcionar a éstos un elevado nivel de seguridad.
2. En virtud de lo establecido en el artículo 11.2 de la Ley 9/2003, de 25 de abril, este capítulo no será de aplicación al transporte por cualquier medio de organismos modificados genéticamente ni a las sustancias y compuestos medicinales de uso humano que consistan en organismos modificados genéticamente o en combinaciones de estos, o que contengan dichos organismos, siempre que su liberación voluntaria, con finalidad distinta a su comercialización, esté autorizada por otras normas comunitarias o por la legislación española dictada para su cumplimiento, siempre que dicha normativa prevea:
a) Una evaluación del riesgo para la salud humana y el medio ambiente, realizada de conformidad con lo establecido en el anexo IV, sin perjuicio de cualesquiera otros requisitos adicionales previstos en la normativa mencionada en este apartado.
b) Una autorización expresa previa a la liberación.
c) Un plan de seguimiento conforme a lo establecido en el anexo V, con vistas a detectar los efectos de los organismos modificados genéticamente sobre la salud humana o el medio ambiente.
d) Los requisitos oportunos del tratamiento de nuevos datos, información al público, datos sobre resultados de la liberación e intercambios de información equivalentes, al menos, a los previstos en la Ley 9/2003, de 25 de abril, y en este reglamento.
No obstante, cuando existan estas disposiciones especiales para las sustancias y compuestos medicinales de uso humano, el órgano competente para su autorización solicitará previamente al Consejo interministerial de organismos modificados genéticamente un informe sobre la evaluación del riesgo para la salud humana y el medio ambiente.
3. La normativa a que se refiere el apartado anterior deberá disponer los procedimientos que garanticen la conformidad de la evaluación del riesgo para la salud y el medio ambiente, y su equivalencia con las disposiciones de la Ley 9/2003, de 25 de abril, y de este reglamento, y deberá hacer referencia a éstas.
4. No será precisa la autorización de liberación voluntaria en los casos relacionados con el examen técnico de variedades modificadas genéticamente cuando las modificaciones genéticas que contengan hayan obtenido, con anterioridad, la autorización de comercialización conforme a lo previsto en la Ley 9/2003, de 25 de abril, y en este reglamento.
5. No se comercializará ningún material derivado de organismos modificados genéticamente que hayan sido liberados de manera voluntaria en los términos de este capítulo, salvo si ésta se realiza de conformidad con lo dispuesto en el capítulo III de este título.
[Bloque 43: #a23]
1. Las personas físicas o jurídicas que se propongan realizar una liberación voluntaria de organismos modificados genéticamente deberán solicitar autorización al órgano competente.
2. La solicitud de autorización deberá contener:
a) Un estudio técnico, que proporcione la información especificada en el anexo V que sea necesaria para llevar a cabo la evaluación del riesgo para la salud humana y el medio ambiente de la liberación voluntaria y, como mínimo:
1.º Información general que incluya la relativa al personal y su formación.
2.º Información relativa a los organismos modificados genéticamente.
3.º Información relativa a las condiciones de liberación y al posible entorno receptor.
4.º Información sobre la interacción de los organismos modificados genéticamente y el medio ambiente.
5.º Un plan de seguimiento, para determinar los efectos de los organismos modificados genéticamente sobre la salud humana y el medio ambiente.
6.º Información sobre el control, los métodos de reparación, el tratamiento de residuos y los planes de actuación en caso de emergencia.
7.º Un resumen del expediente, que se ajustará al modelo establecido en el anexo de la Decisión 2002/813/CE del Consejo, de 3 de octubre de 2002.
b) Una evaluación del riesgo para la salud humana y el medio ambiente y las conclusiones exigidas en la sección D del anexo IV, junto con las referencias bibliográficas e indicadores relativos a los métodos utilizados para llegar a dichas conclusiones. La evaluación del riesgo deberá efectuarse de conformidad con los principios establecidos en el anexo IV de este reglamento, las notas de orientación complementarias contenidas en el anexo de la Decisión 2002/623/CE de la Comisión, de 24 de julio de 2002, y atendiendo a la información facilitada conforme al anexo V de este reglamento.
En dicha evaluación del riesgo se deberá tener debidamente en cuenta los organismos modificados genéticamente que contengan genes que expresen resistencia a los antibióticos utilizados en tratamientos médicos o veterinarios, a fin de identificar y eliminar de forma progresiva en los organismos modificados genéticamente los marcadores de resistencia a los antibióticos que puedan tener efectos adversos para la salud humana y el medio ambiente, conforme a la disposición adicional primera de la Ley 9/2003, de 25 de abril, por la que se establece el régimen jurídico de la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente.
En cada caso, la evaluación del riesgo deberá tener en cuenta los potenciales efectos adversos que puedan tener directa o indirectamente sobre la salud humana o el medio ambiente las transferencias genéticas de organismos modificados genéticamente a otros organismos.
Cualquier transferencia genética de organismos modificados genéticamente a otros organismos deberá someterse, caso por caso, a una evaluación de los potenciales efectos adversos que puedan tener directa o indirectamente sobre la salud humana y el medio ambiente.
3. El titular de la actividad podrá remitirse a datos o resultados de liberaciones efectuadas previamente por otros titulares, siempre que dicha información no sea confidencial o que los titulares precedentes hayan dado su consentimiento por escrito. Igualmente, el titular de la actividad podrá presentar la información complementaria que considere pertinente.
4. El órgano competente podrá aceptar que se presente una única solicitud de autorización cuando el titular de la actividad se proponga realizar liberaciones de un mismo organismo modificado genéticamente o de una combinación de organismos modificados genéticamente en un mismo lugar o en diferentes lugares con la misma finalidad y dentro de un periodo definido.
5. La solicitud de autorización a que se refiere el apartado 2 se presentará con formatos de datos normalizados, cuando existan, con arreglo al Derecho de la Unión Europea.
Se añade el apartado 5 por el art. 2.5 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
[Bloque 44: #a24]
1. Las solicitudes de autorización de liberaciones voluntarias de organismos modificados genéticamente se dirigirán al órgano competente en cada caso, que acusará recibo de ellas.
2. Cuando la competencia corresponda a la Administración General del Estado, la solicitud de autorización se dirigirá al Secretario General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en su calidad de Presidente del Consejo Interministerial de OMG.
Cuando se trate de solicitudes de personas jurídicas, deberán presentarse en todo caso electrónicamente, según lo establecido en el artículo 14.2 de la Ley 39/2015, de 1 de octubre, mientras que cuando se trate de personas físicas podrá realizarse la presentación en cualquiera de los lugares previstos en el artículo 16.4 de la Ley 39/2015, de 1 de octubre.
El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de los de la Comisión Nacional de Bioseguridad dicha solicitud a efectos de lo establecido en el apartado 1 de la disposición adicional segunda de la Ley 9/2003, de 25 de abril.
3. Cuando la competencia corresponda a las comunidades autónomas, el titular de la actividad deberá presentar ante el órgano competente de aquélla la correspondiente solicitud de autorización.
El órgano competente, una vez realizadas las actuaciones de comprobación documental oportunas, remitirá inmediatamente una copia de la solicitud de autorización a la Dirección General de Desarrollo Sostenible del Medio Rural para el cumplimiento por parte de ésta de las obligaciones de información con la Comisión Europea y con los demás Estados miembros.
El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de la Comisión Nacional de Bioseguridad una copia de dicha solicitud para que ésta emita el preceptivo informe de conformidad con lo establecido en el apartado 2 de la disposición adicional segunda de la Ley 9/2003, de 25 de abril, y en el artículo 9 de este reglamento.
Se modifica el apartado 2 por el art. 2.6 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifican los apartados 2 y 3 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 45: #a25]
1. Una vez recibida la solicitud y acusado recibo de su fecha de recepción, el órgano competente examinará que la información, los datos y documentos contenidos en la solicitud son completos y exactos, evaluará los riesgos que representa la liberación y comprobará que todo ello, así como la actividad que se pretende realizar, se ajusta a las disposiciones de la Ley 9/2003, de 25 de abril, y de este reglamento.
La Dirección General de Desarrollo Sostenible del Medio Rural, en el plazo de 30 días a partir de la recepción de la solicitud de autorización, enviará el resumen del expediente contemplado en el artículo 23.2 para su información y traslado a la Comisión Europea y posterior remisión por ésta a los demás Estados miembros. Tras la recepción, en su caso, de las observaciones de la Comisión Europea o de otros Estados miembros, la Dirección General de Desarrollo Sostenible del Medio Rural remitirá éstas a los órganos competentes de las comunidades autónomas y a la Comisión Nacional de Bioseguridad, en los supuestos en que éstas ostenten la competencia para otorgar la autorización de liberación voluntaria de organismos modificados genéticamente de acuerdo con lo dispuesto en la Ley 9/2003, de 25 de abril, y en este reglamento.
2. A efectos de la resolución de la autorización, el órgano competente podrá exigir al responsable de la liberación voluntaria de organismos modificados genéticamente que proporcione cualquier información adicional o realice cuantas pruebas se estimen convenientes, justificando dicha exigencia.
3. El órgano competente podrá consultar a los expertos, instituciones o Administraciones públicas cuando por las características o naturaleza de la actividad que se vaya a desarrollar lo estime conveniente.
4. El órgano competente someterá a información pública, durante un plazo de treinta días, el proyecto de liberación voluntaria, que podrá reducirse hasta un mínimo de veinte días por resolución del órgano competente, por motivos de necesidad. En todo caso, se considera necesidad cuando la actividad esté relacionada con la prevención o contención de agentes infecciosos, procesos o situaciones que puedan tener repercusiones para la salud y para los que se precise actuaciones inmediatas o a corto plazo. Todo ello sin perjuicio de la posible aplicación del procedimiento de la tramitación de urgencia, en los términos del artículo 33 de la Ley 39/2015, de 1 de octubre.
Sin perjuicio de lo establecido en el artículo 48, la información al público deberá incluir el resumen del expediente contemplado en el artículo 23.2, en especial el nombre y dirección del titular de la actividad, la descripción general del organismo modificado genéticamente que va a ser liberado, el lugar y el propósito de la liberación y, por último, el periodo previsto de la liberación.
5. Cuando la competencia corresponda a la Administración General del Estado, el Consejo interministerial de organismos modificados genéticamente solicitará, con carácter previo a la adopción de la resolución, un informe a la Comisión Nacional de Bioseguridad. Se remitirá copia de la solicitud a las comunidades autónomas para que formulen las observaciones que estimen pertinentes en el plazo máximo de diez días.
6. Cuando la competencia corresponda a las comunidades autónomas, el órgano competente correspondiente solicitará, con carácter previo a la adopción de la resolución, un informe a la Comisión Nacional de Bioseguridad.
Se modifican los apartados 4 y 5 por el art. 2.6 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifica el párrafo segundo del apartado 1 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 46: #a26]
1. El órgano competente, una vez analizados los documentos y datos aportados, los resultados de la información pública y, en su caso, los resultados de las consultas, las informaciones adicionales, las observaciones realizadas por otros Estados miembros y el informe de la Comisión Nacional de Bioseguridad, resolverá sobre la liberación voluntaria de organismos modificados genéticamente solicitada, autorizándola o denegándola según se cumplan o no los requisitos establecidos en la Ley 9/2003, de 25 de abril, y en este reglamento. La resolución que autorice la liberación voluntaria impondrá las condiciones necesarias para su realización, deberá ser expresa y notificarse por escrito al titular de la actividad en el plazo de tres meses desde la recepción de la solicitud de autorización.
Para calcular el plazo de los tres meses mencionados anteriormente, no se tendrán en cuenta los períodos de tiempo en que el órgano competente esté a la espera de recibir la información adicional solicitada al titular o bien esté realizando la información pública, que en ningún caso deberá prolongar en más de 30 días el período de los tres meses citados.
2. La Dirección General de Desarrollo Sostenible del Medio Rural informará a la Comisión Europea de las resoluciones adoptadas de acuerdo con lo dispuesto en este artículo, incluidos, en su caso, los motivos por los que se deniega la autorización. Cuando la Administración competente sea la comunidad autónoma, ésta facilitará a la Dirección General de Desarrollo Sostenible del Medio Rural la información necesaria para cumplir dicha obligación, en virtud de lo establecido en la disposición final segunda de la Ley 9/2003, de 25 de abril.
Se modifica el apartado 2 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 47: #a27]
1. El titular de la actividad de liberación está obligado a informar al órgano competente del resultado de la liberación voluntaria de organismos modificados genéticamente en relación con los riesgos para la salud humana y el medio ambiente, y hará constar, en su caso, la intención de proceder a la futura comercialización del organismo liberado o de un producto que lo contenga. Dicha información se proporcionará en los intervalos establecidos en la autorización a la vista de los resultados de la evaluación del riesgo para la salud humana y el medio ambiente y, en el caso de plantas superiores modificadas genéticamente, conforme al modelo que figura en el anexo XI.
2. La Dirección General de Desarrollo Sostenible del Medio Rural informará a la Comisión Europea de los resultados de las liberaciones recibidos con arreglo al apartado anterior en virtud de lo establecido en la disposición final segunda de la Ley 9/2003, de 25 de abril.
Se modifica el apartado 2 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 48: #a28]
1. Cuando se haya adquirido experiencia suficiente en la liberación de determinados organismos modificados genéticamente en ecosistemas específicos y dichos organismos reúnan los criterios que se establecen en el anexo VI, la Dirección General de Desarrollo Sostenible del Medio Rural podrá presentar a la Comisión Europea una propuesta motivada para aplicar a tales tipos de organismos modificados genéticamente procedimientos diferenciados con la finalidad de que la Comisión Europea tome una decisión al respecto.
2. La Dirección General de Desarrollo Sostenible del Medio Rural informará a la Comisión Europea de su decisión de aplicar o no un procedimiento diferenciado establecido en cualquier decisión comunitaria.
Se modifica por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 49: #a29]
1. No obstante lo establecido en los artículos anteriores, podrá presentarse una única solicitud de autorización para efectuar varias liberaciones voluntarias de vegetales modificados genéticamente que se hayan generado a partir de las mismas plantas receptoras cultivadas pero que puedan diferir en cualquiera de las secuencias insertadas o suprimidas o tener las mismas secuencias insertadas o suprimidas pero diferir en el fenotipo.
Igualmente, se podrá presentar una única solicitud de autorización para un programa de desarrollo de un trabajo completo especificado previamente, con una única especie vegetal receptora y un rango especificado de inserciones y supresiones durante varios años en lugares distintos.
2. De acuerdo con la Decisión 94/730/CEE de la Comisión, de 4 de noviembre de 1994, las autorizaciones reguladas en el apartado anterior se concederán mediante el procedimiento simplificado recogido en el anexo VII, siempre que se cumplan las siguientes condiciones:
a) Que la taxonomía y la biología de las plantas receptoras sean bien conocidas.
b) Que se cuente con la información sobre las interacciones de las especies vegetales receptoras en los ecosistemas en los que se programa una liberación agrícola o experimental.
c) Que existan datos sobre la seguridad para la salud humana y para el medio ambiente de las liberaciones experimentales en que estén presentes plantas modificadas genéticamente de las mismas especies vegetales receptoras.
d) Que las secuencias insertadas y los productos de su expresión sean seguros para la salud humana y el medio ambiente en las condiciones de la liberación experimental.
e) Que se hayan caracterizado correctamente las secuencias insertadas.
f) Que todas las secuencias insertadas se integren en el genoma nuclear de la planta.
g) Que todas las liberaciones pertenezcan a un mismo programa de trabajo establecido a priori.
h) Que todas las liberaciones se realicen en un período limitado y fijado previamente.
[Bloque 50: #ciii]
[Bloque 51: #a30]
1. Se entiende por comercialización todo acto que suponga una entrega a terceros, a título oneroso o gratuito, de organismos modificados genéticamente o de productos que los contengan.
2. No se consideran comercialización de organismos modificados genéticamente las siguientes operaciones:
a) El suministro de organismos modificados genéticamente para actividades reguladas en el capítulo I de este título, incluidas las colecciones de cultivos.
b) El suministro de organismos modificados genéticamente para utilizarlos exclusivamente para liberaciones voluntarias que cumplan los requisitos establecidos en el capítulo II del este título.
[Bloque 52: #a31]
1. Lo dispuesto en este capítulo no será de aplicación:
a) Al transporte por cualquier medio de organismos modificados genéticamente.
b) A los organismos modificados genéticamente que sean productos o componentes de productos, tales como las variedades vegetales modificadas genéticamente y los medicamentos de uso humano o veterinario que consistan en organismos modificados genéticamente o en combinaciones de estos, o que contengan dichos organismos, regulados por normas comunitarias distintas a las incorporadas por la Ley 9/2003, de 25 de abril, y este reglamento, siempre que éstas exijan una evaluación específica de los riesgos para el medio ambiente equivalente a la regulada en la citada ley y en este reglamento. Cuando se trate de productos o componentes de productos, tales como las variedades vegetales modificadas genéticamente, estas normas específicas deberán contener, además, requisitos en materia de gestión de riesgo, etiquetado, seguimiento, en su caso, información al público y cláusula de salvaguardia, equivalentes a los previstos en la Ley 9/2003, de 25 de abril, y en este reglamento.
Durante la valoración de las solicitudes de comercialización de organismos modificados genéticamente a que se refiere el párrafo anterior, los órganos competentes para otorgar la autorización, solicitarán previamente al Consejo interministerial de organismos modificados genéticamente un informe sobre la evaluación específica del riesgo ambiental.
2. Hasta que no se adopte la legislación necesaria para garantizar que los requisitos recogidos en el apartado 1.b) sean equivalentes a los que prevé la Ley 9/2003, de 25 de abril, y este reglamento, únicamente se podrán comercializar los organismos modificados genéticamente que sean productos o componentes de productos regulados por normas comunitarias distintas a las incorporadas por la Ley 9/2003, de 25 de abril, o por este reglamento, una vez que su comercialización haya sido autorizada por el órgano competente de la Administración General del Estado de conformidad con este capítulo, sin perjuicio de la obtención de otras autorizaciones que les sean de aplicación.
Redactado el apartado 1.b) conforme a la corrección de errores publicada en BOE núm. 42, de 18 de febrero de 2004. Ref. BOE-A-2004-2974
[Bloque 53: #a32]
1. Las personas físicas o jurídicas que pretendan comercializar por primera vez organismos modificados genéticamente, o una combinación de éstos como productos o componentes de productos, solicitarán autorización al Consejo interministerial de organismos modificados genéticamente y sólo podrán proceder a la comercialización cuando hayan recibido dicha autorización y cumplan las condiciones que estipule la misma.
2. La solicitud de autorización incluirá:
a) Un estudio técnico, que comprenda las informaciones y datos contenidos en los anexos V y VIII, y que deberá tener en cuenta la diversidad de lugares de uso de los organismos modificados genéticamente que sean productos o componentes de un producto e incluir los datos y resultados obtenidos en las liberaciones voluntarias con fines de investigación y desarrollo, sobre las consecuencias de la liberación para la salud humana y el medio ambiente.
b) Una evaluación del riesgo para la salud humana y el medio ambiente y las conclusiones exigidas en la sección D del anexo IV. La evaluación del riesgo deberá efectuarse de conformidad con los principios establecidos en el anexo IV de este reglamento, las notas de orientación complementarias contenidas en el anexo de la Decisión 2002/623/CE de la Comisión, de 24 de julio de 2002, y atendiendo a la información facilitada conforme a los anexos V y VIII de este reglamento.
En dicha evaluación del riesgo se deberá tener debidamente en cuenta los organismos modificados genéticamente que contengan genes que expresen resistencia a los antibióticos utilizados en tratamientos médicos o veterinarios, a fin de identificar y eliminar de forma progresiva en los organismos modificados genéticamente los marcadores de resistencia a los antibióticos que puedan tener efectos adversos para la salud humana y el medio ambiente, conforme a la disposición adicional primera de la Ley 9/2003, de 25 de abril.
En cada caso, la evaluación del riesgo deberá tener en cuenta los potenciales efectos adversos que puedan tener directa o indirectamente sobre la salud humana o el medio ambiente las transferencias genéticas de organismos modificados genéticamente a otros organismos.
c) Las condiciones para la comercialización del producto, incluidas las de uso y manejo.
d) Un plan de seguimiento, de conformidad con el anexo X, con una propuesta de vigencia del plan, que podrá ser distinta del período de duración de la autorización.
e) Una propuesta de etiquetado y de envasado, que deberá cumplir los requisitos establecidos en el anexo VIII. El etiquetado indicará claramente la presencia de organismos modificados genéticamente. En la etiqueta o en la documentación adjunta deberá figurar la frase: «Este producto contiene organismos modificados genéticamente».
f) La propuesta del período de duración de la autorización, que no podrá ser superior a 10 años.
g) La información de que se disponga, en su caso, sobre datos o resultados de otras liberaciones del mismo organismo modificado genéticamente en trámite de autorización o ya efectuadas, tanto por el interesado como por terceras personas, siempre que éstas hayan dado su conformidad por escrito.
h) Un resumen del expediente, que se presentará conforme al modelo establecido en el anexo de la Decisión 2002/812/CE del Consejo, de 3 de octubre de 2002.
3. Deberá solicitarse una nueva autorización para la comercialización de aquellos productos que, aun conteniendo los mismos organismos modificados genéticamente que los incluidos en otros productos ya autorizados, vayan a destinarse a diferente uso.
4. Si, sobre la base de los resultados de liberaciones autorizadas de conformidad con el capítulo II de este título, o en virtud de otros argumentos científicos justificados y sólidos, el solicitante de la autorización considera que la comercialización y el uso de un organismo modificado genéticamente, ya sea como producto o como componente de un producto, no presenta ningún riesgo para la salud humana ni para el medio ambiente, podrá proponer al Consejo interministerial de organismos modificados genéticamente no presentar toda o parte de la información exigida en el apartado B del anexo VIII.
5. La Dirección General de Desarrollo Sostenible del Medio Rural o la Comisión Europea podrán proponer criterios y requisitos de información que deberán cumplir las solicitudes para comercializar determinados tipos de organismos modificados genéticamente como productos o componentes de productos. Dichos criterios y requisitos se adoptarán conforme al procedimiento comunitario establecido al efecto, y sustituirán a lo establecido en el apartado 2 de este artículo, sin perjuicio de la aplicación de las restantes disposiciones de este capítulo exigibles para la obtención de las autorizaciones de comercialización de organismos modificados genéticamente.
6. La solicitud de autorización a que se refiere el apartado 2 se presentará con formatos de datos normalizados, cuando existan, con arreglo al Derecho de la Unión Europea.
Se añade el apartado 6 por el art. 2.7 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifica el apartado 5 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 54: #a33]
1. Las solicitudes de autorización reguladas en este capítulo se dirigirán a la persona titular de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en su calidad de Presidente del Consejo Interministerial de OMG, quien acusará recibo de aquellas.
Cuando se trate de solicitudes de personas jurídicas deberán presentarse en todo caso electrónicamente, según lo establecido en el artículo 14.2 de la Ley 39/2015, de 1 de octubre, mientras que cuando se trate de personas físicas podrá realizarse la presentación en cualquiera de los lugares previstos en el artículo 16.4 de la Ley 39/2015, de 1 de octubre.
2. El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de los de la Comisión Nacional de Bioseguridad dichas solicitudes, a efectos de que el órgano colegiado, previo informe de la citada Comisión, adopte la resolución correspondiente.
3. Por el Consejo interministerial de organismos modificados genéticamente simultáneamente al cumplimiento del trámite de informe previsto en el apartado anterior, se dará traslado de las solicitudes a las comunidades autónomas para que formulen las observaciones que estimen pertinentes en el plazo de 10 días.
Se modifica el apartado 1 por el art. 2.7 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifican los apartados 1 y 2 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 55: #a34]
1. Una vez recibida la solicitud de autorización y acusado recibo de ella, el Consejo interministerial de organismos modificados genéticamente remitirá inmediatamente el resumen del expediente a que se refiere el artículo 32.2.h) a las autoridades competentes de los demás Estados miembros y a la Comisión Europea.
2. El Consejo interministerial de organismos modificados genéticamente comprobará sin demora si la información aportada reúne los requisitos del artículo 32.2 y podrá, en su caso, pedir al titular de la solicitud de autorización que proporcione la información adicional que considere necesaria.
Una vez haya comprobado que la solicitud de autorización reúne los requisitos del artículo 32.2, remitirá, a más tardar cuando envíe el informe de evaluación previsto en el artículo 15 de la Ley 9/2003, de 25 de abril, una copia de la solicitud a la Comisión Europea, que la remitirá a las autoridades competentes de los demás Estados miembros y la pondrá a disposición del público en los términos previstos en la normativa comunitaria.
3. En el plazo de tres meses a partir de la recepción de la solicitud, el Consejo interministerial de organismos modificados preparará un informe de evaluación del riesgo, con arreglo a las directrices establecidas en el anexo IX, y lo remitirá al titular de la solicitud de autorización. El informe de evaluación indicará si los organismos modificados genéticamente de que se trate deben o no comercializarse y en qué condiciones.
Tras la recepción del informe de evaluación, el titular de la solicitud podrá retirar el expediente. Esto no imposibilita una posterior solicitud de autorización a una autoridad competente de otro Estado miembro.
4. Si el informe de evaluación indica que los organismos modificados genéticamente de que se trate pueden comercializarse, el Consejo interministerial de organismos modificados genéticamente remitirá dicho informe de evaluación a la Comisión Europea junto con la información adicional presentada por el titular de la solicitud de autorización y cualquier otra información que haya servido de base a su informe.
Una vez recibida la documentación a la que hace referencia el párrafo anterior, la Comisión Europea la remitirá a las autoridades competentes de los demás Estados miembros dentro de un plazo de 30 días a partir de la fecha de su recepción de la citada documentación.
5. Si el informe de evaluación indica que los organismos modificados genéticamente de que se trate no deben comercializarse, el Consejo interministerial de organismos modificados genéticamente remitirá dicho informe de evaluación a la Comisión Europea junto con la información adicional presentada por el titular de la solicitud y cualquier otra información que haya servido de base a su informe.
Dicha remisión se realizará, como mínimo 15 días después del envío del informe de evaluación al titular de la solicitud de autorización previsto en el apartado 3, y como máximo 105 días después de la presentación de la solicitud de autorización por el citado titular al Consejo interministerial de organismos modificados genéticamente.
Una vez recibida la documentación a la que hacen referencia los párrafos anteriores, la Comisión Europea la remitirá a las autoridades competentes de los demás Estados miembros dentro de un plazo de 30 días a partir de la fecha de su recepción.
6. A la hora de computar el plazo de tres meses mencionado en el apartado 3, no se tendrán en cuenta los períodos de tiempo en los que el Consejo interministerial de organismos modificados genéticamente haya estado a la espera de la información adicional que hubiera solicitado justificadamente al titular de la solicitud de autorización.
[Bloque 56: #a35]
1. Una vez recibido el informe de evaluación y la documentación correspondiente a una solicitud de comercialización presentada en otro Estado miembro por medio de la Comisión Europea, el Consejo interministerial de organismos modificados genéticamente dispondrá de un plazo de 60 días para solicitar motivadamente más información, formular observaciones o presentar objeciones motivadas a la comercialización de los organismos modificados genéticamente de que se trate, y de un plazo de 105 días, desde la recepción de dicho informe, para estudiar junto con la Comisión Europea y el resto de los Estados miembros los asuntos pendientes y llegar a un acuerdo. Para la realización de estas actividades, el Consejo interministerial de organismos modificados genéticamente recabará informe de la Comisión Nacional de Bioseguridad.
2. Las respuestas a las solicitudes de información, las observaciones o las objeciones previstas en el apartado anterior se enviarán a la Comisión Europea, que las trasmitirá de inmediato a los demás Estados miembros.
3. Los períodos en los que se haya estado esperando recibir más información del interesado no se contarán a los efectos del cálculo del día en que venza el plazo para alcanzar un acuerdo.
4. Durante el procedimiento de autorización de comercialización para cultivo de un determinado OMG, previsto en la parte C de la Directiva 2001/18/CE, del Parlamento Europeo y del Consejo, de 12 de marzo de 2001 y en el Reglamento (CE) 1829/2003 del Parlamento Europeo y del Consejo, de 22 de septiembre de 2003, se podrán:
a) Adoptar medidas que den lugar a una restricción en el cultivo de ese OMG en el territorio nacional, de conformidad con los procedimientos previstos en los apartados 1 a 4 del artículo 26 ter de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001.
b) Adoptar medidas para reintegrar el territorio nacional en las autorizaciones previstas en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, y del Reglamento (CE) 1829/2003 del Parlamento Europeo y del Consejo de 22 de septiembre de 2003, de conformidad con el apartado 5 del artículo 26 ter de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, en el caso de que se hubiera producido una restricción en el cultivo de acuerdo con lo previsto en el subapartado anterior.
Con carácter previo a la adopción de dichas medidas, el Consejo Interministerial recabará informe a la Comisión Nacional de Bioseguridad y consultará a las comunidades autónomas.
Se añade el apartado 4 por el art. 2.8 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
[Bloque 57: #a36]
1. La autorización de comercialización de organismos modificados genéticamente sólo podrá otorgarse cuando se haya autorizado previamente la liberación voluntaria sin fines comerciales de dichos organismos, o se haya realizado la evaluación del riesgo para la salud humana y el medio ambiente a que se refiere el artículo 4.a), conforme a lo dispuesto en la Ley 9/2003, de 25 de abril, y en este reglamento. En todo caso, los productos deberán cumplir las normas vigentes sobre comercialización de productos.
2. El Consejo interministerial de organismos modificados genéticamente concederá la autorización por escrito, la transmitirá al titular de la solicitud e informará de ella a los demás Estados miembros y a la Comisión Europea, en el plazo de 30 días, si una vez transcurrido el plazo de 60 días contemplado en el artículo 34 no se han recibido objeciones justificadas de un Estado miembro o de la Comisión Europea o si se han resuelto los posibles asuntos pendientes en el plazo de 105 días.
3. Si, por el contrario, se hubiesen formulado objeciones y no se llegase a un acuerdo entre los Estados miembros, la autorización se someterá al procedimiento que se establezca en la normativa comunitaria y dicha autorización no podrá ser concedida sin la previa aprobación de la Comisión Europea. Cuando en dicho procedimiento se haya adoptado una decisión favorable, el Consejo interministerial de organismos modificados genéticamente concederá la autorización por escrito y la transmitirá al titular de la solicitud e informará a los demás Estados miembros y a la Comisión Europea, en el plazo de 30 días desde la publicación o notificación de dicha decisión.
4. Las autorizaciones otorgadas por cualquier Estado miembro habilitarán para que el organismo modificado genéticamente o el producto que lo contenga pueda ser comercializado en España, siempre que dichas autorizaciones se hayan otorgado de acuerdo con las disposiciones que incorporen a los respectivos derechos nacionales las normas comunitarias sobre esta materia y se respeten estrictamente las condiciones establecidas en las respectivas autorizaciones y, en particular, las condiciones relativas al entorno y a las áreas geográficas.
5. Conforme a lo establecido en la disposición adicional primera del real decreto que aprueba este reglamento, las autorizaciones de comercialización se incluirán en el Registro central, y serán accesibles al público.
[Bloque 58: #a37]
La resolución de autorización de comercialización de organismos modificados genéticamente deberá contener:
a) El alcance de la autorización, incluida la identidad de los organismos modificados genéticamente que se van a comercializar como productos o componentes de productos, y su identificador único.
b) El plazo de validez de la autorización, que no podrá ser superior a 10 años contados desde la fecha de autorización.
c) Las condiciones de comercialización del producto, incluidas las condiciones específicas de uso, manejo y embalaje de los organismos modificados genéticamente como productos o componentes de productos y los requisitos para la protección de determinados ecosistemas, entornos o áreas geográficas particulares.
d) La obligación del interesado de facilitar muestras de control al Consejo interministerial de organismos modificados genéticamente cuando éste se lo solicite.
e) Los requisitos de etiquetado conforme a lo establecido en el anexo VIII. El etiquetado indicará claramente la presencia de organismos modificados genéticamente. La mención: «Este producto contiene organismos modificados genéticamente» aparecerá en la etiqueta o en un documento que acompañe a los productos que contengan organismos modificados genéticamente.
f) Los requisitos y el plan de seguimiento previstos en el artículo 42, realizados de conformidad con el anexo X, incluidas las obligaciones de información a la Comisión Europea y a los demás Estados miembros y el plazo para el plan de seguimiento. Cuando proceda, se identificarán las obligaciones de las personas que comercialicen el producto o de sus usuarios, así como la información que se considere adecuada sobre la localización de los organismos modificados genéticamente cultivados.
La resolución de autorización especificará caso por caso los requisitos, procedimientos y obligaciones del plan de seguimiento.
Cualquier medida similar establecida por otras normas comunitarias específicas o por la legislación española dictada para su cumplimiento deberá tener en cuenta los planes de seguimiento contemplados en la Ley 9/2003, de 25 de abril, y en este reglamento. Para tal fin, dichas medidas se pondrán en conocimiento de la Comisión Nacional de Bioseguridad con antelación suficiente antes de su aprobación.
[Bloque 59: #a38]
1. La autorización se concederá por un plazo máximo de 10 años a partir de la fecha de la autorización.
2. A efectos de la aprobación de un organismo modificado genéticamente o de su progenie con vistas únicamente a la comercialización de sus semillas de conformidad con las disposiciones comunitarias aplicables, el período de la primera autorización concluirá a más tardar 10 años a partir de la fecha de la primera inscripción en el Registro de variedades comerciales previsto en la Ley 11/1971, de 30 de marzo, de semillas y plantas de vivero, de la primera variedad vegetal que contenga organismos modificados genéticamente.
En el caso de materiales forestales de reproducción, el período de la primera autorización concluirá a más tardar 10 años a partir de la fecha de la primera inscripción del material de base con organismo modificado genéticamente en el Catálogo nacional de materiales de base, regulado de conformidad con la Directiva 1999/105/CE del Consejo, de 22 de diciembre de 1999, sobre la comercialización de materiales forestales de reproducción.
[Bloque 60: #a39]
1. De acuerdo con lo establecido en el artículo 16.4 y en la disposición transitoria segunda de la Ley 9/2003, de 25 de abril, las autorizaciones concedidas de conformidad con este capítulo y aquellas concedidas antes del 17 de octubre de 2002 deberán renovarse.
2. Las personas físicas y jurídicas que pretendan renovar su autorización de comercialización deberán presentar una solicitud de renovación en un plazo máximo de nueve meses antes de que expire la autorización obtenida de acuerdo con este capítulo o antes del 17 de octubre de 2006 si fueron concedidas antes del 17 de octubre de 2002.
3. La solicitud de renovación incluirá lo siguiente:
a) Un ejemplar de la autorización de comercialización de los organismos modificados genéticamente.
b) Un informe de los resultados del seguimiento realizado con arreglo al artículo 42. En caso de las autorizaciones obtenidas antes del 17 de octubre de 2002, dicho informe se presentará cuando se hubiera efectuado el seguimiento.
c) Toda nueva información de que se disponga en relación con los riesgos que el producto entraña para la salud humana o el medio ambiente.
d) Cuando proceda, una propuesta para modificar o complementar las condiciones de la autorización inicial, es decir, las relativas al futuro seguimiento y el plazo de la autorización.
4. Tras presentar la solicitud para renovar la autorización, el titular de la solicitud podrá seguir comercializando los organismos modificados genéticamente con arreglo a las condiciones establecidas en dicha autorización hasta que se resuelva definitivamente su renovación.
[Bloque 61: #a40]
1. Las solicitudes de renovación de las autorizaciones reguladas en este capítulo se dirigirán al Secretario General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en su calidad de Presidente del Consejo Interministerial de OMG, quien acusará recibo de aquellas.
Cuando se trate de solicitudes de personas jurídicas deberán presentarse en todo caso electrónicamente, según lo establecido en el artículo 14.2 de la Ley 39/2015, de 1 de octubre, mientras que cuando se trate de personas físicas podrá realizarse la presentación en cualquiera de los lugares previstos en el artículo 16.4 de la Ley 39/2015, de 1 de octubre.
2. El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial del organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de los de la Comisión Nacional de Bioseguridad dichas solicitudes, a efectos de que el Consejo interministerial del organismos modificados genéticamente, previo informe de la citada Comisión, adopte la resolución correspondiente.
Se modifica el apartado 1 por el art. 2.9 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifica por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 62: #a41]
1. Una vez recibida la solicitud de renovación de la autorización y acusado recibo de ella, el Consejo interministerial de organismos modificados genéticamente, previo informe de la Comisión Nacional de Bioseguridad, comprobará que la solicitud de renovación de autorización se ajusta al artículo 39.3 y realizará un informe de evaluación con arreglo a las directrices establecidas en el anexo IX. Este informe de evaluación indicará si el organismo modificado genéticamente debe seguir o no en el mercado y en qué condiciones.
El Consejo interministerial de organismos modificados genéticamente procederá sin demora a remitir un ejemplar de la solicitud de renovación, así como su informe de evaluación, a la Comisión Europea, que, en un plazo de 30 días a partir de la fecha de recepción, los remitirá a las autoridades competentes de los demás Estados miembros. El informe de evaluación se remitirá también al titular de la solicitud.
2. Una vez recibido el informe de evaluación, los Estados miembros y la Comisión Europea tienen un plazo de 60 días para solicitar más información, formular observaciones, o presentar objeciones motivadas a la comercialización de los organismos modificados genéticamente de que se trate. Las respuestas a las solicitudes de información, las observaciones o las objeciones se remitirán a la Comisión Europea, que las comunicará inmediatamente a los demás Estados miembros. Los Estados miembros y la Comisión Europea podrán debatir los aspectos pendientes con objeto de alcanzar un acuerdo, para lo que dispondrán de un plazo adicional de 15 días.
3. El Consejo interministerial de organismos modificados genéticamente concederá la renovación de la autorización por escrito y la transmitirá al interesado e informará de ella a los demás Estados miembros y a la Comisión Europea, en el plazo de 30 días, si una vez transcurrido el plazo de 60 días desde que se facilitó el informe de evaluación, no se han recibido objeciones justificadas de un Estado miembro o de la Comisión Europea, o si se han resuelto los posibles asuntos pendientes en el plazo de 75 días desde la fecha de recepción del informe de evaluación.
4. La validez de la autorización renovada no debe superar los 10 años. No obstante, este plazo podrá reducirse o ampliarse según convenga por motivos específicos, cuando no se hubieran recibido objeciones justificadas de un Estado miembro o de la Comisión Europea.
[Bloque 63: #a42]
1. Tras la comercialización de organismos modificados genéticamente como productos o componentes de productos, la persona física o jurídica autorizada para comercializar el producto, de conformidad con lo que establece el artículo 32, velará por que el seguimiento y la presentación de informes sobre aquéllos se lleven a cabo con arreglo a las condiciones especificadas en la autorización. Basándose en dichos informes, de conformidad con los términos de la autorización y en el marco del plan de seguimiento especificado en ella, el Consejo interministerial de organismos modificados genéticamente podrá, previo informe de la Comisión Nacional de Bioseguridad, adaptar el plan de seguimiento al finalizar el primer período de seguimiento, de conformidad con el anexo X.
2. El Consejo interministerial de organismos modificados genéticamente enviará informes de dicho seguimiento a la Comisión Europea y a las autoridades competentes de los demás Estados miembros.
[Bloque 64: #a43]
1. No se podrá prohibir, restringir o impedir la comercialización de organismos modificados genéticamente que hayan sido autorizados por otros Estados miembros siempre que estas autorizaciones se hayan otorgado de acuerdo con las disposiciones que incorporen a los respectivos derechos nacionales las normas de las Comunidades Europeas sobre esta materia y se respeten estrictamente las condiciones establecidas en las respectivas autorizaciones.
2. No obstante, el Consejo interministerial de organismos modificados genéticamente podrá restringir o suspender temporalmente el uso y la venta en su territorio de un organismo modificado genéticamente que sea un producto o un componente de producto debidamente autorizado por otro Estado miembro, cuando con posterioridad a su autorización disponga de nuevas informaciones de las que se deduzca que representa un riesgo para la salud humana y el medio ambiente. Dicha información debe afectar a la evaluación del riesgo para la salud humana y el medio ambiente o tratarse de una nueva valoración de la información existente a tenor de conocimientos científicos nuevos o adicionales.
En el caso de que, a tenor de la información prevista en el párrafo anterior, se conociera la existencia de riesgo grave, se aplicarán medidas de emergencia, tales como la suspensión o el cese de la comercialización.
En los supuestos contemplados en los párrafos anteriores de este apartado, se informará al público.
3. En los casos previstos en el apartado anterior, el Consejo interministerial de organismos modificados genéticamente informará inmediatamente a la Comisión Europea y a los demás Estados miembros sobre las acciones adoptadas, exponiendo los motivos de su decisión y facilitando la nueva valoración de la evaluación del riesgo para la salud humana y el medio ambiente, indicando si deben modificarse, y en qué forma, las condiciones de la autorización o si debe ponerse fin a esta última y, cuando proceda, la información nueva o adicional en que se base su decisión.
[Bloque 65: #a44]
1. Sin perjuicio de lo establecido en el artículo 46.2, si antes de conceder la autorización el Consejo interministerial de organismos modificados genéticamente recibe información que pueda repercutir en el riesgo de la comercialización de los organismos modificados genéticamente para la salud humana o el medio ambiente, transmitirá inmediatamente la información a la Comisión Europea y a las autoridades competentes de los demás Estados miembros. En este supuesto, se podrán aplicar los plazos establecidos para la resolución de los asuntos pendientes en los artículos 35.1 y 41.3, según se trate de una solicitud de autorización o de una renovación de ésta.
2. Cuando se haya accedido a la información señalada en al apartado anterior después de conceder la autorización, el Consejo interministerial de organismos modificados genéticamente, previo informe de la Comisión Nacional de Bioseguridad, enviará a la Comisión Europea su informe de evaluación dentro de un plazo de 60 días a partir de la fecha de recepción de la nueva información, indicando si se deben modificar las condiciones de la autorización y cómo, o si ésta se debe revocar. La Comisión Europea transmitirá dicho informe a los demás Estados miembros, dentro de los 30 días siguientes al de su recepción.
3. Si dentro del plazo de 60 días a partir de la fecha de remisión de la nueva información no se presenta objeción motivada por algún Estado miembro o por la Comisión Europea, o si en el plazo de 75 días se resuelven los asuntos pendientes, el Consejo interministerial de organismos modificados genéticamente que redactó el informe modificará la autorización en el sentido propuesto, enviará la autorización modificada al titular de la comercialización e informará de ello a los demás Estados miembros y a la Comisión Europea dentro de un plazo de 30 días.
[Bloque 66: #a45]
1. Los operadores, definidos en el artículo 4.e), que comercialicen productos que contengan o estén compuestos por organismos modificados genéticamente en cualquier fase de su producción o distribución conservarán y transmitirán por escrito al operador que reciba el producto la siguiente información:
a) La mención de que el producto contiene o está compuesto por organismos modificados genéticamente.
b) El identificador o identificadores únicos asignados a dichos organismos modificados genéticamente con arreglo al procedimiento comunitario que se establezca al respecto.
2. En el caso de los productos que estén compuestos por mezclas de organismos modificados genéticamente o las contengan, destinados a utilizarse única y directamente como alimentos o piensos, o a ser procesados, la información recogida en el apartado 1.b) podrá sustituirse por una declaración de uso del operador, junto con una lista de los identificadores únicos de todos los organismos modificados genéticamente que hayan sido utilizados para constituir la mezcla.
3. Los operadores conservarán la información especificada en el apartado 1 durante los cinco años posteriores a cada transacción comercial con el fin de saber de qué operador proceden y a qué operador han sido suministrados los productos.
4. Para los productos mencionados en los apartados 1 y 2 anteriores, los operadores garantizarán que:
a) En el caso de los productos preenvasados la etiqueta contendrá la siguiente indicación: «Este producto contiene organismos modificados genéticamente», o bien: «Este producto contiene (nombre de los organismos) modificado genéticamente».
b) En el caso de productos no preenvasados ofrecidos al consumidor final, la indicación: «Este producto contiene organismos modificados genéticamente», o bien: «Este producto contiene (nombre de los organismos) modificado genéticamente», constará en la presentación del producto o en la documentación que acompaña al producto.
5. Los apartados anteriores no se aplicarán a las trazas de organismos modificados genéticamente que estén presentes en una proporción no superior a los umbrales fijados en el Reglamento (CE) n.º 1829/2003 del Parlamento Europeo y del Consejo, de 22 de septiembre de 2003, o por otras normas específicas de la Unión Europea, siempre y cuando dichas trazas sean accidentales o técnicamente inevitables.
[Bloque 67: #civ]
[Bloque 68: #a46]
1. Cuando, con posterioridad a la presentación de las comunicaciones, de las solicitudes de autorización o de su otorgamiento reguladas en los capítulos I, II y III de este título, se disponga de nuevos datos respecto de los riesgos que la actividad pueda representar para la salud humana y el medio ambiente, su titular está obligado a informar inmediatamente al órgano competente, a revisar las informaciones y los requisitos especificados en la comunicación, solicitud o autorización y a adoptar las medidas necesarias para proteger la salud humana y el medio ambiente.
A efectos de este apartado, cuando la competencia para recibir la información corresponda a la Administración General del Estado, la comunicación se dirigirá al Secretario General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en su calidad de Presidente del Consejo Interministerial de OMG. Cuando se trate de la remisión de información por personas jurídicas deberá presentarse en todo caso electrónicamente, según lo establecido en el artículo 14.2 de la Ley 39/2015, de 1 de octubre, mientras que cuando se trate de personas físicas podrá realizarse la presentación en cualquiera de los lugares previstos en el artículo 16.4 de la Ley 39/2015, de 1 de octubre.
El Director General de Desarrollo Sostenible del Medio Rural, a través del secretario del Consejo interministerial de organismos modificados genéticamente, pondrá en conocimiento de sus miembros y de los de la Comisión Nacional de Bioseguridad dicha comunicación.
2. Cuando el órgano competente disponga de informaciones de las que se deduzca que la actividad puede representar riesgos superiores a los previstos, exigirá al titular la modificación de las condiciones de ejecución, su suspensión o la finalización de la actividad, e informará de ello al público.
A tales efectos, cuando la competencia corresponda a la Administración General del Estado, el Consejo interministerial de organismos modificados genéticamente adoptará la correspondiente resolución, previo informe de la Comisión Nacional de Bioseguridad.
Se modifica el apartado 1 por el art. 2.10 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifican los párrafos segundo y tercero del apartado 1 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 69: #a47]
1. Las obligaciones previstas en el artículo anterior serán también aplicables si una modificación en la utilización confinada o en la liberación voluntaria pudiera tener consecuencias respecto a los riesgos para la salud humana o para el medio ambiente. En este supuesto se debe informar al órgano competente antes de que se produzca la modificación o cuando se conozca el cambio no intencionado.
2. Para los supuestos de comercialización de organismos modificados genéticamente, se estará a lo dispuesto en el artículo anterior y en el artículo 44.
[Bloque 70: #a48]
1. En el ejercicio de las competencias atribuidas en la Ley 9/2003, de 25 de abril, y en este reglamento, las Administraciones competentes garantizarán que se respetan y protegen los derechos de propiedad intelectual y, en su caso, industrial, así como la normativa sobre libertad de acceso de información en materia de medio ambiente.
2. Las autoridades competentes no comunicarán a terceros la información confidencial notificada o intercambiada con arreglo al presente reglamento.
3. Las personas físicas o jurídicas que pretendan o se propongan realizar las actividades reguladas en los artículos 13, 23 y 32 de la Ley 9/2003, de 25 de abril, y en este reglamento, podrán presentar a la autoridad una petición de tratamiento confidencial acompañada de una justificación verificable, de conformidad con lo dispuesto en los próximos apartados.
4. En el caso de las comunicaciones o solicitudes de autorización de actividades de utilización confinada, la autoridad competente, previa consulta con la persona física o jurídica que presente la comunicación decidirá la información que deberá mantenerse en secreto y le informará de su decisión. En ningún caso deberá mantenerse en secreto la información siguiente:
a) Características generales del organismo modificado genéticamente, nombre y dirección del notificante y lugar de utilización.
b) Tipo de utilización confinada y medidas de confinamiento.
c) Evaluación de los efectos previsibles y, en particular, de cualquier efecto nocivo para la salud y para el medio ambiente.
Si, por cualquier razón, la persona física o jurídica retira la comunicación o solicitud, la autoridad competente, deberá respetar el carácter confidencial de la información facilitada.
5. En el caso de las solicitudes de autorización de liberación voluntaria y comercialización, la autoridad competente, evaluará la petición de confidencialidad presentada y podrá otorgar tratamiento confidencial únicamente a los elementos de información que se indican a continuación, previa justificación verificable, siempre que la persona o entidad solicitante demuestre que su revelación podría conllevar un perjuicio importante para sus intereses:
a) El proceso de fabricación o elaboración, incluido el método y aspectos innovadores de este, así como otras especificaciones técnicas e industriales inherentes a dicho proceso o método, a excepción de la información que sea pertinente para la evaluación de la seguridad.
b) Las relaciones comerciales entre la persona o entidad productora o importadora y la solicitante o titular de la autorización, en su caso.
c) La información comercial que revele las fuentes, la cuota de mercado o la estrategia comercial de la persona o entidad solicitante.
d) Información sobre la secuencia del ADN, excepto en el caso de secuencias utilizadas a efectos de detección, identificación y cuantificación del evento de transformación.
e) Pautas y estrategias de reproducción.
La autoridad decidirá, previa consulta a las personas físicas o jurídicas que presenten la solicitud de autorización, qué información debe recibir el tratamiento confidencial y les informará de su decisión.
Se adoptarán las medidas necesarias para garantizar que no se hace pública la información confidencial notificada o intercambiada con arreglo a la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, sobre la liberación intencional en el medio ambiente de organismos modificados genéticamente y por la que se deroga la Directiva 90/220/CEE del Consejo, y en particular en caso de que proceda consultar al comité científico conforme al artículo 28 de la citada directiva.
Asimismo, se aplicará lo dispuesto en el artículo 25.6 de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001 para el tratamiento de los datos personales y el acceso a la información.
No obstante, la autoridad competente podrá divulgar la información a la que se refiere en los párrafos anteriores cuando sea indispensable una intervención urgente para proteger la salud humana, la sanidad o el medio ambiente, como en situaciones de emergencia. También se hará pública la información que forme parte de las conclusiones de las contribuciones científicas aportadas por el comité o comités científicos pertinentes, o las conclusiones de los informes de evaluación y que se relacionen con efectos previsibles en la salud humana, la sanidad animal y el medioambiente, de conformidad con lo establecido en el artículo 39 quater del Reglamento (CE) n.º 178/2002 del Parlamento Europeo y del Consejo, de 28 de enero de 2002, por el que se establecen los principios y los requisitos generales de la legislación alimentaria, se crea la Autoridad Europea de Seguridad Alimentaria y se fijan procedimientos relativos a la seguridad alimentaria.
En el caso de que las personas físicas o jurídicas retiren la solicitud de autorización, se respetará la confidencialidad otorgada por la autoridad competente de conformidad con lo dispuesto en el presente artículo. Si se retira antes de que la autoridad haya tomado una decisión sobre la petición de confidencialidad no se hará pública la información para la que había pedido tratamiento confidencial.
Se modifica por el art. 2.8 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
[Bloque 71: #a49]
Conforme a lo establecido en la disposición adicional única del real decreto que aprueba este reglamento y sin perjuicio de lo establecido en el artículo anterior, se pondrá a disposición del público la información relativa a las autorizaciones de utilización confinada, liberaciones voluntarias con fines distintos a su comercialización y la comercialización de organismos modificados genéticamente. Igualmente, se pondrá a disposición del público la relación de los organismos modificados genéticamente cuya comercialización haya sido autorizada o rechazada como productos o componentes de productos, los informes de evaluación, los resultados de los controles sobre comercialización y los informes de la Comisión Nacional de Bioseguridad. Se especificará para cada producto tanto los organismos modificados genéticamente que contenga como sus usos.
[Bloque 72: #a50]
1. Sin perjuicio de lo establecido en el artículo 45, los órganos competentes de las Administraciones públicas garantizarán que, en cualquier fase de la comercialización de organismos modificados genéticamente, se cumplan las condiciones de etiquetado y envasado establecidas en la autorización.
2. Así mismo los citados órganos garantizarán que los operadores apliquen los umbrales mínimos establecidos por la Comisión Europea, por debajo de los cuales no necesitarán etiquetarse los productos respecto de los cuales no puedan excluirse rastros accidentales o técnicamente inevitables de organismos modificados genéticamente autorizados.
3. Los organismos modificados genéticamente que deban facilitarse para las operaciones previstas en el artículo 30.2 estarán sujetos a los requisitos adecuados de etiquetado de conformidad con lo establecido en el anexo VIII, con el fin de proporcionar información clara en una etiqueta o en un documento de acompañamiento sobre la presencia de organismos modificados genéticamente. Para ello, figurarán en una etiqueta o en un documento de acompañamiento las palabras «Este producto contiene organismos modificados genéticamente».
[Bloque 73: #a51]
1. Al final de cada año natural, la Secretaría General de Agricultura y Alimentación deberá remitir a la Comisión Europea:
a) Un informe sintético sobre las utilizaciones confinadas de los tipos 3 y 4, autorizadas durante dicho año, conforme al artículo 18 y siguientes, en el que conste la descripción, la finalidad y los riesgos de la utilización confinada de que se trate.
b) Una relación de los organismos modificados genéticamente liberados en su territorio y una lista de las solicitudes de autorización rechazadas.
2. De acuerdo con lo establecido en la disposición final cuarta de la Ley 9/2003, de 25 de abril, cada tres años la Secretaría General de Agricultura y Alimentación elaborará un informe, que se enviará a la Comisión Europea y se hará público, sobre la situación en el Reino de España y la experiencia adquirida en materia de organismos modificados genéticamente.
3. La Secretaría General de Agricultura y Alimentación, en los términos establecidos en la Ley 9/2003, de 25 de abril, en este reglamento y en la normativa europea vigente, intercambiará información con la Comisión Europea acerca de la experiencia adquirida en materia de prevención de los riesgos asociados a la utilización confinada, liberación voluntaria y comercialización de organismos modificados genéticamente. Asimismo, dicho intercambio de información incluirá también la aplicación del artículo 30.2 de este reglamento, la evaluación del riesgo para la salud humana y el medio ambiente, el seguimiento y la consulta e información al público.
4. La Secretaría General de Agricultura y Alimentación informará a la Comisión Europea y a los demás Estados miembros sobre las acciones, sanciones y medidas aplicadas para poner fin a las utilizaciones confinadas, liberaciones voluntarias y comercializaciones de organismos modificados genéticamente, que se hayan efectuado sin la preceptiva autorización.
5. A efecto de cumplimentar lo previsto en los apartados anteriores, las comunidades autónomas facilitarán a la Secretaría General de Agricultura y Alimentación los datos necesarios para cumplir las obligaciones de información a la Comisión Europea.
6. La información a que se refieren los apartados 1 a 4 se pondrá también en conocimiento de los Ministerios para la Transición Ecológica, de Sanidad, Consumo y Bienestar Social, y de Ciencia, Innovación y Universidades, así como de cualesquiera otros Departamentos ministeriales u organismos de la Administración General del Estado con competencias en esta materia.
7. De conformidad con lo establecido en la disposición final segunda de la Ley 9/2003, de 25 de abril, el Ministerio de Agricultura, Pesca y Alimentación pondrá a disposición de las comunidades autónomas la información a que se refieren los apartados 1 a 4 anteriores.
Asimismo, la Secretaría General de Agricultura y Alimentación informará trimestralmente al órgano competente de las comunidades autónomas sobre las autorizaciones otorgadas por el Consejo interministerial de organismos modificados genéticamente respecto de actividades que se desarrollen en el ámbito geográfico de cada comunidad autónoma.
Se modifica por el art. 2.11 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifica el párrafo primero del apartado 1 por el art. 19.6 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
[Bloque 74: #a52]
Para el cómputo de los períodos a que se hace referencia en los artículos 16, 19, 26.1, 34.3 y 35.1, no serán tenidos en cuenta los períodos durante los cuales el órgano competente esté a la espera de la información adicional que pueda haber solicitado, o esté llevando a cabo, en su caso, un periodo de información pública con arreglo a los artículos 16.2 y 25.4.
[Bloque 75: #a53]
La falta de resolución expresa por el órgano competente para otorgar las autorizaciones reguladas en la Ley 9/2003, de 25 de abril, y en este reglamento producirá efectos desestimatorios.
[Bloque 76: #tiii]
[Bloque 77: #ci-3]
[Bloque 78: #a54]
Los titulares de las actividades a que se refiere la Ley 9/2003, de 25 de abril, y este reglamento estarán obligados a prestar toda la colaboración a las autoridades competentes a fin de permitirles realizar los exámenes, controles, toma de muestras y recogida de información necesaria para el cumplimiento de su misión.
[Bloque 79: #a55]
Los funcionarios y empleados públicos que realicen las labores de inspección en las actividades reguladas en la Ley 9/2003, de 25 de abril, y en este reglamento tendrán el carácter de agentes de la autoridad.
A dichos efectos, los funcionarios que realicen labores de inspección y control estarán facultados para requerir cuanta información y documentación sea precisa para el ejercicio de sus funciones y para acceder a las superficies, medios e instalaciones en las que se realicen actividades reguladas en la Ley 9/2003, de 25 de abril, y en este reglamento, salvo que tuvieran la consideración legal de domicilio, en cuyo caso la labor inspectora deberá ajustarse a las normas que garantizan su inviolabilidad.
En todo caso, las actividades de inspección y control realizadas y el resultado de éstas deberán consignarse en las correspondientes actas extendidas por los funcionarios que realizaran estas actuaciones.
Las actas tendrán valor probatorio respecto de los hechos reflejados en ellas, sin perjuicio de las pruebas que en defensa de sus derechos puedan aportar los interesados.
[Bloque 80: #a56]
1. Son obligaciones de los inspectores:
a) Realizar las actuaciones inspectoras y de control precisas para llegar al convencimiento razonable de que las actividades, instalaciones y demás elementos objeto de aquéllas cumplen los requisitos exigidos por la normativa europea y nacional.
b) Elaborar las correspondientes actas de inspección.
c) Expedir los certificados que correspondan de acuerdo con la normativa vigente, siempre y cuando el objeto de la inspección sea acreedor del certificado, y, en todo caso, siempre que el interesado lo solicite.
2. Los inspectores, en el ejercicio de sus funciones, deberán portar un documento personal acreditativo, expedido por la Administración competente, que deberá presentarse a efectos de realizar sus funciones.
3. En el ejercicio de sus funciones deberá observarse el deber de respeto y consideración debido a los interesados, y se adoptarán todas las medidas necesarias para garantizar la protección de la intimidad de las personas y las precisas para no dificultar más allá de lo que fuera indispensable el normal funcionamiento de las actividades objeto de control e inspección.
Los inspectores están, así mismo, obligados a guardar secreto profesional sobre las actividades realizadas y a respetar la normativa en materia de incompatibilidades.
[Bloque 81: #cii-3]
[Bloque 82: #a57]
Tendrán la consideración de infracciones a lo regulado en este reglamento las establecidas en el artículo 34 de la Ley 9/2003, de 25 de abril, sin perjuicio, en su caso, de las posibles responsabilidades civiles y penales derivadas de las infracciones cometidas.
[Bloque 83: #a58]
1. Serán responsables de las infracciones las personas físicas o jurídicas siguientes:
a) En el caso de que se realicen actividades para las que se requieren autorización en virtud del título II de este reglamento, sin la autorización correspondiente, las personas físicas o jurídicas que las realizasen.
b) En el caso de incumplimiento de las condiciones de la autorización de actividades previstas en el título II de este reglamento, los titulares de dichas autorizaciones.
c) En el caso de que se realicen actividades de utilización confinada de organismos modificados genéticamente que no requieran autorización administrativa, las personas físicas o jurídicas que se propongan realizar o realicen dichas actividades.
d) En el caso de la comercialización de productos que contengan o estén compuestos por organismos modificados genéticamente, los operadores.
2. Cuando los daños causados al medio ambiente o a la salud de las personas se produzcan por acumulación de actividades debidas a diferentes personas, el órgano competente podrá imputar individualmente esta responsabilidad y sus efectos económicos.
[Bloque 84: #ciii-2]
[Bloque 85: #a59]
Las infracciones a lo dispuesto en la Ley 9/2003, de 25 de abril, y en este reglamento serán sancionadas de conformidad con lo previsto en el artículo 35.1 de la citada ley.
[Bloque 86: #a60]
1. Corresponde a los órganos competentes de las comunidades autónomas la imposición de las sanciones por la comisión de infracciones cometidas en la realización de las actividades reguladas en la Ley 9/2003, de 25 de abril, y en este reglamento, con las excepciones reguladas en los apartados siguientes.
2. La Administración General del Estado será competente para imponer las sanciones por la comisión de las infracciones relacionadas con la realización de las actividades a que se refieren la letra c) del apartado 1, y las letras a), b) y c) del apartado 2, del artículo 3 de la Ley 9/2003, de 25 de abril, con sujeción a lo previsto en los apartados siguientes.
3. La imposición de las sanciones por la comisión de infracciones relacionadas con las actividades reguladas en el artículo 3.2.a) de la Ley 9/2003, de 25 de abril, así como las relacionadas con la importación y exportación de OMG y de los productos que los contengan, cuando la comisión de las infracciones implique riesgo para la salud humana y para el control sanitario del medio ambiente, corresponderá:
a) A la persona titular de la Dirección General de Salud Pública del Ministerio de Sanidad en el caso de las infracciones leves.
b) A la persona titular de la Secretaría de Estado de Sanidad del Ministerio de Sanidad, en el caso de las infracciones graves.
c) A la persona titular del Ministerio de Sanidad, en el caso de las infracciones muy graves.
4. La competencia para la imposición de sanciones por la comisión de infracciones relacionadas con la realización de los programas de investigación a que se refiere el artículo 3.2.b) de la Ley 9/2003, de 25 de abril, corresponderá:
a) A la persona titular de la Dirección General de Planificación de la Investigación del Ministerio de Ciencia e Innovación, en el caso de las infracciones leves.
b) A la persona titular de la Secretaría General de Investigación del Ministerio de Ciencia e Innovación, en el caso de las infracciones graves.
c) A la persona titular del Ministerio de Ciencia e Innovación, en el caso de las infracciones muy graves.
5. La imposición de las sanciones por la comisión de infracciones cometidas en los supuestos de examen técnico a los que se refiere el artículo 3.2.c) de la ley anteriormente citada, así como las realizadas en materia de importación y exportación de semillas y plantas de vivero que incorporen o contengan OMG, sin perjuicio de lo dispuesto en el apartado 3 de este artículo, corresponderá:
a) A la persona titular de la Dirección General de Producciones y Mercados Agrarios del Ministerio de Agricultura, Pesca y Alimentación, en el caso de las infracciones leves.
b) A la persona titular de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en el caso de las infracciones graves.
c) A la persona titular del Ministerio de Agricultura, Pesca y Alimentación, en el caso de las infracciones muy graves.
6. La imposición de sanciones por la comisión de infracciones cometidas en relación con la importación y exportación de OMG y de los productos que los contengan para su utilización en actividades de biorremediación o en otras distintas de las referidas en los apartados 3, 4 y 5, siempre que no supongan riesgo para la salud humana o para el control sanitario del medio ambiente, corresponderá:
a) A la persona titular de la Dirección General de Producciones y Mercados Agrarios del Ministerio de Agricultura, Pesca y Alimentación, en el caso de las infracciones leves.
b) A la persona titular de la Secretaría General de Agricultura y Alimentación del Ministerio de Agricultura, Pesca y Alimentación, en el caso de las infracciones graves.
c) A la persona titular del Ministerio de Agricultura, Pesca y Alimentación, en el caso de las infracciones muy graves.
7. Los órganos sancionadores a los que se refiere este artículo tendrán en cuenta en sus resoluciones lo establecido en el artículo 35.4 de la Ley 9/2003, de 25 de abril.
Se modifica por el art. 2.9 del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Se modifica por el art. 2.12 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se modifican los apartados 3 a 6 por el art. 2.4 del Real Decreto 191/2013, de 15 de marzo. Ref. BOE-A-2013-3365.
Se modifican los apartados 5 y 6 por el art. 19.7 del Real Decreto 367/2010, de 26 de marzo. Ref. BOE-A-2010-5037.
Redactado el párrafo primero del apartado 5 conforme a la corrección de errores publicada en BOE núm. 42, de 18 de febrero de 2004. Ref. BOE-A-2004-2974
[Bloque 87: #a61]
Las sanciones se impondrán atendiendo a las circunstancias del responsable, su grado de culpa, la reiteración, participación y beneficio obtenido y grado de incidencia o riesgo objetivo de daño grave a la salud humana, el medio ambiente o los recursos naturales.
Cuando la cuantía de la multa resulte inferior al beneficio obtenido por la comisión de la infracción, la sanción será aumentada, como mínimo, hasta el doble del importe en que se haya beneficiado el infractor.
[Bloque 88: #a62]
Cuando, antes de iniciarse un procedimiento sancionador, la Administración competente comprobase que la actividad se realiza sin la correspondiente autorización o sin haberse comunicado o cuando pueda causar daño grave a la salud humana o al medio ambiente, podrá acordar el precinto o cierre de la instalación o de la parte de la instalación donde se realiza dicha actividad y, en su caso, proceder a la inmovilización o decomiso de los organismos modificados genéticamente o de los productos que los contengan. El órgano competente para iniciar el correspondiente procedimiento sancionador o el instructor del expediente deberán decidir sobre su continuidad o su levantamiento en el plazo de 15 días a partir de aquel en el que se hayan acordado las citadas medidas.
[Bloque 89: #a63]
Cuando se haya iniciado un procedimiento sancionador, el órgano competente podrá adoptar alguna o algunas de las medidas provisionales siguientes:
a) Cierre temporal, parcial o total, suspensión o paralización de las instalaciones.
b) Suspensión temporal de la autorización para el ejercicio de la actividad.
c) Inmovilización de los organismos modificados genéticamente o de los productos que los contengan
d) Cualesquiera otras medidas de corrección, seguridad o control que impidan la continuidad en la producción del daño.
[Bloque 90: #a64]
1. Sin perjuicio de las sanciones que procedan, los responsables de actividades infractoras quedarán obligados a reponer las cosas al estado que tuvieran antes de la infracción, así como a abonar la correspondiente indemnización por los daños y perjuicios causados, cuyo importe será fijado por la Administración que en cada caso resulte competente, sin perjuicio de la competencia correspondiente a jueces y tribunales.
Cuando los daños fueran de difícil evaluación se aplicarán, conjunta o separadamente, los siguientes criterios: coste teórico de la restitución y reposición, valor de los bienes dañados, coste del proyecto o actividad causante del daño y beneficio obtenido con la actividad infractora.
2. Si, una vez finalizado el procedimiento sancionador y transcurridos los plazos señalados en el correspondiente requerimiento, el infractor no procediera a la reparación o restitución exigida en el apartado anterior, el órgano competente podrá acordar la imposición de multas coercitivas. La cuantía de cada una de las multas no superará un tercio de la multa fijada para la infracción cometida.
3. Asimismo, el órgano competente podrá proceder a la ejecución subsidiaria por cuenta del infractor y a su costa.
[Bloque 91: #a65]
1. La potestad sancionadora en el ámbito de aplicación de la Ley 9/2003, de 25 de abril, y de este reglamento, así como la prescripción de las infracciones y sanciones, se regirán por lo dispuesto en la Ley 39/2015, de 1 de octubre.
2. Concluido un procedimiento sancionador, la resolución se pondrá en conocimiento de otros órganos de la misma o de las distintas Administraciones públicas con competencias en materia de organismos modificados genéticamente.
Se modifica por el art. 2.13 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
[Bloque 92: #da]
1. En el caso de que se cultiven OMG en terrenos fronterizos con otro u otros Estados miembros en el que esta actividad esté prohibida, se adoptarán medidas adecuadas para evitar una posible contaminación transfronteriza.
2. Las medidas se adoptarán mediante orden u órdenes del Ministro de Agricultura, Pesca y Alimentación, siendo preceptivo disponer previamente de un informe de evaluación de riesgo de la Comisión Nacional de Bioseguridad y de la correspondiente audiencia a la comunidad o comunidades autónomas en cuyo ámbito territorial vayan a surtir efectos.
3. Estas medidas serán proporcionales, no discriminatorias, basadas en el principio de prevención y cautela, y el principio caso por caso. No será obligatoria su adopción si en el informe de evaluación de riesgo se determina que no son necesarias por razones geográficas especiales.
4. La orden u órdenes a que se refiere el apartado2 se adoptarán una vez se haya concedido la autorización o renovación de la autorización de cultivo de ese OMG en la Unión Europea, y se haya hecho efectiva la prohibición en el Estado miembro fronterizo o Estados miembros fronterizos.
5. En un plazo no superior a60 días desde su entrada en vigor, se procederá a la comunicación de las normas adoptadas a la Comisión Europea.
6. El seguimiento, vigilancia y control y, en su caso sanción, de las medidas a que se refiere esta disposición corresponderá a las comunidades autónomas.
Se modifica el apartado 2 por el art. 2.14 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Se añade, con efectos desde el día 3 de abril de 2017, por el art. único del Real Decreto 364/2017, de 17 de abril. Ref. BOE-A-2017-4242
Texto añadido, publicado el 18/04/2017, en vigor a partir del 19/04/2017.
[Bloque 93: #ani]
Este anexo describe en términos generales los elementos que se deberán tener en cuenta y el procedimiento que se deberá seguir para realizar la evaluación del riesgo para la salud humana y el medio ambiente a la que hace referencia el artículo 12 de este reglamento. Como complemento a este anexo deberán utilizarse las notas de orientación establecidas en la Decisión 2000/608/CE de la Comisión, de 27 de septiembre de 2000, y cumplir cualesquiera otras disposiciones comunitarias que se aprueben en la materia, bien sean de aplicación directa o una vez que se produzca su incorporación al ordenamiento interno.
A. ELEMENTOS DE EVALUACIÓN
1. Los siguientes efectos deberán considerarse potencialmente nocivos:
Enfermedades que afecten a las personas, incluidos los efectos alérgicos o tóxicos.
Enfermedades que afecten a los animales o a los vegetales.
Efectos deletéreos debidos a la imposibilidad de tratar una enfermedad o de realizar una profilaxis eficaz.
Efectos deletéreos debidos al establecimiento o a la diseminación en el medio ambiente.
Efectos deletéreos debidos a la transferencia natural de material genético insertado a otros organismos.
2. La evaluación mencionada en el artículo 12 deberá fundarse en lo siguiente:
a) Identificación de cualquier efecto potencialmente nocivo y, en particular, aquellos que estén relacionados con:
El organismo receptor.
El material genético insertado procedente del organismo donante.
El vector.
El organismo donante (si se utiliza durante la operación).
El organismo modificado genéticamente resultante.
b) Características de la actividad.
c) Gravedad de los efectos potencialmente nocivos.
d) Probabilidad de que se produzcan los efectos potencialmente nocivos.
B. PROCEDIMIENTO
1. El primer paso en el proceso de evaluación debe consistir en identificar las propiedades nocivas del organismo receptor y, en su caso, del donante, así como cualesquiera propiedades nocivas relacionadas con el vector o con el material introducido, incluidas las alteraciones de las propiedades iniciales del receptor.
2. De manera general, se considerarán apropiados para su inclusión en el tipo 1, tal como queda definido en el artículo 12, únicamente los organismos modificados genéticamente que presenten las siguientes características:
a) Que sea poco probable que el receptor u organismo de origen cause enfermedad en seres humanos, animales o plantas (únicamente animales y plantas pertenecientes al medio ambiente que podría verse expuesto).
b) Que la naturaleza del vector y del material incorporado sean tales que no transmitan al organismo modificado genéticamente un fenotipo que pueda causar enfermedad en seres humanos, animales o plantas (únicamente animales y plantas pertenecientes al medio ambiente que podría verse expuesto), ni efectos deletéreos en el medio ambiente.
c) Que sea poco probable que el organismo modificado genéticamente cause enfermedad en seres humanos, animales o plantas (únicamente animales y plantas pertenecientes al medio ambiente que podría verse expuesto) y que sea poco probable que tenga efectos nocivos sobre el medio ambiente.
3. Para tomar conocimiento de las informaciones necesarias a la puesta en práctica de este proceso, el interesado podrá tener en cuenta en primer lugar la legislación comunitaria existente, en particular la Directiva 90/679/CEE del Consejo, de 26 de noviembre de 1990, así como la normativa que la incorpora al ordenamiento jurídico español, en concreto el Real Decreto 664/1997, de 12 de marzo, sobre la protección de los trabajadores contra riesgos relacionados con la exposición a agentes biológicos durante el trabajo. También podrán tomar en consideración sistemas de clasificación nacionales o internacionales y sus versiones actualizadas conforme a los nuevos conocimientos científicos y el progreso técnico.
Estos sistemas se refieren a organismos naturales y, por consiguiente, se basan normalmente en la capacidad de los organismos para causar enfermedades en seres humanos, animales o plantas y en la gravedad y transmisibilidad de la enfermedad que pueden provocar. El Real Decreto 664/1997, de 12 de marzo, clasifica los organismos, en tanto que agentes biológicos, en cuatro tipos de riesgo en función de sus efectos potenciales en un adulto sano. Dichos tipos de riesgo pueden servir de orientación para la clasificación de las actividades de utilización confinada en los cuatro tipos de riesgo mencionados en el apartado 1 del artículo 12. El interesado también puede tener en cuenta sistemas de clasificación relativos a elementos patógenos vegetales y animales. Los sistemas de clasificación mencionados sólo proporcionan una indicación provisional del tipo de riesgo de la actividad y de las correspondientes medidas de confinamiento y control necesarias.
4. El proceso de identificación de los riesgos, realizado con arreglo a los apartados anteriores de este anexo, debe llevar a la determinación del nivel de riesgo asociado con los organismos modificados genéticamente.
5. A continuación, debe realizarse la selección de las medidas de control y otras medidas de protección con arreglo al nivel de riesgo asociado con los organismos modificados genéticamente, teniendo también en cuenta:
a) Las características del medio ambiente que pueda quedar expuesto a los organismos modificados genéticamente (por ejemplo, la presencia en éste de fauna y flora conocidas que puedan verse afectadas negativamente por los microorganismos empleados en la actividad de utilización confinada).
b) Las características de la actividad (naturaleza, magnitud, etc.).
c) Cualesquiera operaciones no normalizadas (por ejemplo, inoculación de animales con organismos modificados genéticamente, equipo que puede generar aerosoles).
La consideración de los párrafos a) a c) para la actividad de que se trate puede incrementar, reducir o mantener constante el nivel de riesgo asociado con los organismos modificados genéticamente según se establece en el apartado 6.
6. El análisis efectuado conforme a los apartados anteriores conducirá finalmente a la asignación de la actividad a uno de los tipos que se describen en el apartado 1 del artículo 12.
7. La clasificación final de la utilización confinada se confirmará revisando la evaluación contemplada en el artículo 12.
[Bloque 94: #anii]
Principios generales
1. En los cuadros figuran los requisitos habituales mínimos, así como las medidas necesarias para cada grado de confinamiento.
El confinamiento también puede garantizarse mediante el uso de buenas prácticas de trabajo, formación, equipo de confinamiento y diseño particular de las instalaciones. En todas las actividades en que intervengan organismos modificados genéticamente, se aplicarán los principios de las buenas prácticas microbiológicas, así como los principios fundamentales siguientes de seguridad y de higiene en el lugar de trabajo:
a) Mantener la exposición del lugar de trabajo y del medio ambiente a cualquier organismo modificado genéticamente al nivel más bajo posible en la práctica.
b) Aplicar medidas de control industrial en la fuente y, de ser necesario, completar éstas con vestimenta y equipo personal de protección adecuados.
c) Comprobar y mantener de forma adecuada las medidas y equipos de control.
d) Verificar, cuando proceda, la presencia de organismos de proceso viables fuera del confinamiento físico primario.
e) Proporcionar al personal la formación adecuada.
f) Crear comités y subcomités de seguridad biológica, si es preciso.
g) Formular y aplicar códigos de práctica locales para la seguridad del personal, según las necesidades.
h) Si procede, disponer señales de riesgo biológico.
i) Establecer instalaciones de limpieza y descontaminación para el personal.
j) Llevar los correspondientes registros.
k) Prohibir que se coma, beba, fume, se empleen cosméticos o se almacenen alimentos para el consumo humano en la zona de trabajo.
l) Prohibir pipetear con la boca.
m) Establecer, si procede, protocolos de trabajo por escrito con el fin de garantizar la seguridad.
n) Tener a disposición desinfectantes y procedimientos específicos de desinfección en caso de que organismos modificados genéticamente se hayan esparcido.
ñ) Disponer en caso necesario de un lugar de almacenamiento de total seguridad para equipo y materiales de laboratorio contaminados.
2. Los títulos de los cuadros son indicativos.
En el cuadro 1 A figuran los requisitos mínimos para las actividades de laboratorio.
El cuadro I B recoge las adiciones y modificaciones del cuadro I A para las actividades en invernaderos o semilleros con organismos modificados genéticamente.
El cuadro I C recoge las adiciones y modificaciones del cuadro I A para las actividades con animales en las que se empleen organismos modificados genéticamente.
En el cuadro II figuran los requisitos mínimos para las actividades distintas de las de laboratorio.
En algunos casos particulares puede resultar necesario aplicar una combinación de medidas, de los cuadros 1 A y II, correspondientes al mismo grado.
En algunos casos los usuarios podrán, previo acuerdo del órgano competente, no aplicar una especificación en un determinado grado de confinamiento o bien combinar especificaciones de dos grados diferentes.
En estos cuadros el término «facultativa» significa que el usuario podrá aplicar estas medidas en cada caso concreto, en función de la evaluación del riesgo contemplada en el apartado 1 del artículo 12.
3. En aras de la claridad de los requisitos, al aplicar este anexo las Administraciones competentes podrán incorporar además en los cuadros siguientes los principios generales de los apartados 1 y 2.
|
Especificaciones |
Grado de confinamiento |
||||
|---|---|---|---|---|---|
|
1 |
2 |
3 |
4 |
||
|
1 |
Dependencias del laboratorio1 |
No exigida |
No exigida |
Exigida |
Exigida |
|
2 |
Laboratorio: hermético para efectuar una fumigación |
No exigida |
No exigida |
Exigida |
Exigida |
|
3 |
Existencia de una entrada y salida independientes |
No exigida |
No exigida |
Exigida |
Exigida |
|
Equipo |
|||||
|
4 |
Superficies resistentes a ácidos, álcalis, disolventes, desinfectantes y agentes de descontaminación y de fácil limpieza |
Exigida (mesa) |
Exigida (mesa) |
Exigida (mesa y suelo) |
Exigida (mesa, suelo, techo y paredes) |
|
5 |
Acceso al laboratorio a través de una exclusa2 |
No exigida |
No exigida |
Opcional |
Exigida |
|
6 |
Presión negativa respecto a la presión del medio ambiente inmediato |
No exigida |
No exigida |
Exigida con excepción de3 |
Exigida |
|
7 |
Aire de entrada y salida del laboratorio tratado con filtros HEPA |
No exigida |
No exigida |
Exigida: (HEPA)4: aire de salida con excepción de3 |
Exigida: (HEPA)5: aire de entrada y de salida |
|
8 |
Recinto o campana de seguridad microbiológica |
No exigida |
Opcional |
Exigida |
Exigida |
|
9 |
Autoclave |
In situ |
En el edificio |
En las dependencias del laboratorio6 |
En el laboratorio = con dos extremos |
|
Normas de trabajo |
|||||
|
10 |
Acceso restringido |
No exigida |
Exigida |
Exigida |
Exigida |
|
11 |
Señalización de un peligro biológico en la puerta |
No exigida |
Exigida |
Exigida |
Exigida |
|
12 |
Medidas específicas para el control de la formación y difusión de aerosoles |
No exigida |
Exigida: minimizar |
Exigida: evitar |
Exigida: evitar |
|
13 |
Ducha |
No exigida |
No exigida |
Opcional |
Exigida |
|
14 |
Indumentaria de protección |
Indumentaria de protección adecuada |
Indumentaria de protección adecuada |
Indumentaria y calzado de protección adecuados |
Cambio completo de ropa y calzado antes de entrar y salir |
|
15 |
Guantes |
No exigida |
Opcional |
Exigida |
Exigida |
|
18 |
Control eficaz de los vectores (por ejemplo, roedores e insectos) |
Opcional |
Exigida |
Exigida |
Exigida |
|
Residuos |
|||||
|
19 |
Inactivación de los organismos modificados genéticamente en los efluentes de lavabos, desagües y duchas o efluentes similares |
No exigida |
No exigida |
Opcional |
Exigida |
|
20 |
Inactivación de los organismos modificados genéticamente en el material contaminado y en los residuos |
Opcional |
Exigida |
Exigida |
Exigida |
|
Otras medidas |
|||||
|
21 |
Almacenamiento del equipo en el propio laboratorio |
No exigida |
No exigida |
Opcional |
Exigida |
|
23 |
Una ventana de observación o similar para ver a los ocupantes |
Opcional |
Opcional |
Opcional |
Exigida |
1 Aislamiento: el laboratorio se encuentra separado de otras zonas del mismo edilicio o en un edilicio separado.
2 Exclusa: la entrada debe efectuarse a través de una esclusa aislada del laboratorio. El lado limpio de la esclusa ha de estar separado del lado restringido mediante unas instalaciones con vestuarios o duchas y preferiblemente con puertas con cerraduras dependientes.
3 Las actividades en que la transmisión no se realiza por vía aérea.
4 HEPA: Filtro absoluto (High Efficiency Particulate Air).
5 En los locales en los que se manipulen virus que no sean retenidos por los filtros HEPA, serán necesarios otros requisitos relativos al aire de salida.
6 Se permite el transporte seguro de material al autoclave que se encuentre fuera del laboratorio mediante procedimientos validados y con un nivel de protección equivalente.
Los términos «invernadero» y «semillero» se refieren a estructuras con paredes, techo y suelo diseñadas y empleadas principalmente para cultivar plantas en un entorno protegido y controlado.
Se aplicarán todas las normas recogidas en el cuadro 1 A con las adiciones o modificaciones siguientes:
|
Especificaciones |
Grado de confinamiento |
||||
|---|---|---|---|---|---|
|
1 |
2 |
3 |
4 |
||
|
Edificio |
|||||
|
1 |
Invernadero: estructura permanente1 |
No exigida |
Exigida |
Exigida |
Exigida |
|
Equipo |
|||||
|
3 |
Entrada a través de una esclusa con dos puertas con cerradura dependiente |
No exigida |
Opcional |
Opcional |
Exigida |
|
4 |
Control de la escorrentía de agua contaminada |
Opcional |
Minimizar la escorrentía2 |
Evitar la escorrentía |
Evitar la escorrentía |
|
Normas de trabajo |
|||||
|
6 |
Medidas para controlar las especies no deseadas (insectos y otros artrópodos, roedores, etc.) |
Exigida |
Exigida |
Exigida |
Exigida |
|
7 |
Procedimientos para evitar la diseminación de organismos modificados genéticamente durante el transporte de material vivo entre el invernadero o semillero, la estructura protectora y el laboratorio |
Minimizar la diseminación |
Minimizar la diseminación |
Evitar la diseminación |
Evitar la diseminación |
1 El invernadero será una estructura permanente con cubierta continua e impermeable, situada en un lugar cuya pendiente permita evitar la entrada de la escorrentía de aguas superficiales y provista de puertas de cierre automático.
2 Cuando pueda haber transmisión por el suelo.
Se aplicarán todas las normas recogidas en el cuadro I A con las adiciones o modificaciones siguientes
|
Especificaciones |
Grado de confinamiento |
||||
|---|---|---|---|---|---|
|
1 |
2 |
3 |
4 |
||
|
Instalaciones |
|||||
|
1 |
Aislamiento de la unidad de animales1 |
Opcional |
Exigida |
Exigida |
Exigida |
|
2 |
Locales de animales2 separados mediante puertas bloqueables |
Opcional |
Exigida |
Exigida |
Exigida |
|
3 |
Locales de animales diseñados para la descontaminación material (jaulas, etc.), impermeable y fácil de lavar |
Opcional |
Opcional |
Exigida |
Exigida |
|
4 |
Suelo y paredes fáciles de lavar |
Opcional |
Exigida (suelo) |
Exigida (sucio y paredes) |
Exigida (sucio y paredes) |
|
5 |
Confinamiento de los animales en receptáculos adecuados como jaulas, corrales o cajas |
Opcional |
Opcional |
Opcional |
Opcional |
|
6 |
Filtros en las cajas de aislamiento o habitaciones aisladas3 |
No exigida |
Opcional |
Exigida |
Exigida |
1 Unidad de animales: edificio o zona separada de un edificio que disponga de locales y otras zonas como vestuarios, duchas, autoclaves, almacén de alimentos. etc.
2 Locales de animales: locales que habitualmente se empleen para alojar animales de reserva, cría o experimentación o para realizar pequeñas intervenciones quirúrgicas.
3 Cajas de aislamiento: cajas transparentes en las que pueden introducirse animales, dentro o fuera de la jaula; para animales grandes sería más apropiado utilizar habitaciones aisladas.
|
Especificaciones |
Grado de confinamiento |
||||
|---|---|---|---|---|---|
|
1 |
2 |
3 |
4 |
||
|
Disposiciones generales |
|||||
|
1 |
Los organismos viables deben mantenerse en un sistema que separe el proceso del entorno (sistema cerrado) |
Opcional |
Exigida |
Exigida |
Exigida |
|
2 |
Control de los gases de escape del sistema cerrado |
No exigida |
Exigida, minimizando la liberación |
Exigida, evitando la liberación |
Exigida, evitando la liberación |
|
3 |
Control de aerosoles durante la toma de muestras, la introducción de material en un sistema cerrado o la transferencia de material a otro sistema cerrado |
Opcional |
Exigida, minimizando la liberación |
Exigida, evitando la liberación |
Exigida. Evitando la liberación |
|
4 |
Inactivación del líquido de cultivo en masa antes de extraerlo del sistema cerrado |
Opcional |
Exigida, con medios validados |
Exigida, con medios validados |
Exigida, con medios validados |
|
5 |
Sistemas de cierre diseñados para minimizar o evitar la liberación |
Ningún requisito específico |
Minimizar la liberación |
Evitar la liberación |
Evitar la liberación |
|
6 |
Zona controlada con capacidad para contener el vertido de todo el contenido del sistema cerrado. |
Opcional |
Opcional |
Exigida |
Exigida |
|
7 |
Zona controlada hermética para fumigación |
No exigida |
Opcional |
Opcional |
Exigida |
|
Equipo |
|||||
|
8 |
Entrada a través de esclusa |
No exigida |
No exigida |
Opcional |
Exigida |
|
9 |
Superficies resistentes al agua y a los ácidos, álcalis, disolventes, desinfectantes y agentes de descontaminación, y fáciles de limpiar |
Exigida (mesa, sí la hay) |
Exigida (mesa, si la hay) |
Exigida (mesa, si la hay, y suelo) |
Exigida (mesa, suelo, techo y paredes) |
|
10 |
Medidas específicas para ventilar adecuadamente la zona controlada y de este modo minimizar la contaminación atmosférica |
Opcional |
Opcional |
Opcional |
Exigida |
|
11 |
Zona controlada con presión negativa respecto a la presión circundante |
No exigida |
No exigida |
Opcional |
Exigida |
|
12 |
Tratamiento del aire de salida y entrada de la zona filtrado con filtros HEPA |
No exigida |
No exigida |
Exigida (aire de salida; facultativa para el aire de entrada) |
Exigida (arre de entrada y de salida) |
|
Normas de trabajo |
|||||
|
13 |
Sistemas cerrados situados en una zona controlada |
No exigida |
Opcional |
Exigida |
Exigida |
|
14 |
Acceso restringido exclusivamente al personal autorizado |
No exigida |
Exigida |
Exigida |
Exigida |
|
15 |
Obligación de indicar el peligro biológico |
No exigida |
Exigida |
Exigida |
Exigida |
|
17 |
El personal deberá ducharse antes de abandonar la zona controlada |
No exigida |
No exigida |
Opcional |
Exigida |
|
18 |
Indumentaria de protección para el personal |
Exigida (indumentaria de trabajo) |
Exigida (indumentaria de trabajo) |
Exigida |
Cambio total de indumentaria antes de entrar y de salir |
|
Residuos |
|||||
|
22 |
Inactivación de los organismos modificados genéticamente en los efluentes de lavabos y duchas o efluentes similares |
No exigida |
No exigida |
Opcional |
Exigida |
|
23 |
Inactivación de los organismos modificados genéticamente en el material contaminado y los residuos, incluidos los organismos modificados genéticamente presentes en el efluente de trabajo antes del vertido final |
Opcional |
Exigida, con medios validados |
Exigida, con medios validados |
Exigida, con medios validados |
[Bloque 95: #aniii]
Información exigida para la comunicación a que se refiere el artículo 14.1:
1.° Nombre del usuario o usuarios, incluidos los responsables de la supervisión y de la seguridad.
2.° Información sobre la formación profesional y titulación de las personas responsables de la supervisión y de la seguridad.
3.° Datos relativos a todos los comités o subcomités biológicos.
4.° Dirección y descripción general de los locales.
5.° Descripción de la naturaleza del trabajo que se vaya a realizar.
6.° Tipo de utilización confinada.
7.° Únicamente para las utilizaciones confinadas del tipo 1, resumen de la evaluación del riesgo mencionada en el artículo 12 e información sobre gestión de los residuos.
Información exigida para la comunicación a que se refiere el artículo 14.2:
1.° Fecha de presentación de la comunicación a que se refiere el artículo 14.1.
2.° Nombre de las personas responsables de la supervisión y de la seguridad, e información sobre su formación profesional y titulación.
3.° Organismos receptores, donantes y/o parentales y, si procede, sistemas hospedador-vector utilizados.
4.° Procedencia y funciones proyectadas de los materiales genéticos empleados en las modificaciones.
5.° Identidad y características de los organismos modificados genéticamente.
6.° Finalidad de la utilización confinada, incluidos los resultados esperados.
7.° Cantidades aproximadas de cultivos que se vayan a utilizar.
8.° Descripción de las medidas de confinamiento y protección que vayan a aplicarse, incluida la información relativa a la gestión de los residuos, incluyendo los residuos producidos, su tratamiento y su forma y destino finales.
9.° Resumen de la evaluación del riesgo a que se refiere el artículo 12.
10.° Información necesaria para que el órgano competente pueda evaluar los planes de emergencia sanitaria y de vigilancia epidemiológica y medioambiental elaborados de conformidad con el artículo 20.
Información exigida para la comunicación a que se refiere el artículo 14.3:
a) Fecha de presentación de la comunicación mencionada en el artículo 14.1,
Nombre de los responsables de la supervisión y la seguridad e información sobre formación profesional y titulación.
b) Organismos receptores o parentales que vayan a emplearse.
Sistemas hospedador-vector que vayan a emplearse (si procede).
Procedencia y funciones proyectadas de los materiales genéticos empleados en las modificaciones.
Identidad y características del organismo modificado genéticamente.
Cantidades de cultivos que vayan a utilizarse.
c) Descripción de las medidas de confinamiento y otras medidas protectoras que vayan a aplicarse, incluida la información relativa a la gestión de los residuos incluidos el tipo y la forma de los residuos que vayan a producirse, su tratamiento y su forma y destino finales.
Finalidad de la utilización confinada, incluidos los resultados esperados.
Descripción de las partes de la instalación.
d) Información sobre prevención de accidentes y planes de emergencia sanitaria y de vigilancia epidemiológica y medioambiental, si procede:
Riesgos específicos debidos al emplazamiento de la instalación.
Medidas preventivas aplicadas, tales como equipos de seguridad, sistemas de alarma y métodos de confinamiento.
Procedimientos y planes de comprobación de la eficacia permanente de las medidas de confinamiento.
Descripción de la información suministrada a los trabajadores.
Información necesaria para que el órgano competente pueda evaluar los planes de emergencia sanitaria y de vigilancia epidemiológica y medioambiental, elaborados de conformidad con el artículo 20.
e) Un ejemplar de la evaluación del riesgo a que hace referencia el artículo 12.
[Bloque 96: #aniv]
Este anexo describe de modo general el objetivo que debe lograrse, los elementos que deben considerarse y los principios generales y la metodología que deben seguirse para llevar a cabo la evaluación del riesgo para la salud humana y el medio ambiente exigible para realizar actividades de liberación voluntaria y comercialización de organismos modificados genéticamente. Como complemento a este anexo deberán utilizarse las notas de orientación establecidas en la Decisión 2002/623/CE de la Comisión, de 24 de julio de 2002, y cumplir cualesquiera otras disposiciones comunitarias que se aprueben en la materia, bien sean de aplicación directa o una vez que se produzca su incorporación al ordenamiento interno.
Con objeto de contribuir a una comprensión común de los términos «directos, indirectos, inmediatos y diferidos» al aplicar este anexo, sin perjuicio de una posterior orientación a este respecto y en particular en lo que se refiere al alcance en el que los efectos indirectos puedan y deban tenerse en cuenta, se describen estos términos del siguiente modo:
a) Los «efectos directos» hacen referencia a los principales efectos en la salud humana o el medio ambiente que son consecuencia del propio organismo modificado genéticamente y no de una cadena de acontecimientos causal.
b) Los «efectos indirectos» hacen referencia a los efectos en la salud humana o el medio ambiente que son consecuencia de una cadena de acontecimientos causal, a través de mecanismos tales como interacciones con otros organismos, transferencia del material genético o cambios en el uso o la gestión.
Es probable que las observaciones de efectos indirectos tarden en ser observados.
c) Los «efectos inmediatos» hacen referencia a los efectos en la salud humana o el medio ambiente que se observan durante el periodo de la liberación de los organismos modificados genéticamente. Los efectos inmediatos pueden ser directos o indirectos.
d) Los «efectos diferidos» hacen referencia a los efectos en la salud humana o el medio ambiente que no se observan durante el periodo de la liberación de los organismos modificados genéticamente pero que se manifiestan como efectos directos o indirectos bien en una fase posterior, bien una vez concluida la liberación en cuestión.
Un principio general para la evaluación del riesgo para la salud humana y el medio ambiente deberá consistir también en la necesidad de realizar un análisis de los «efectos acumulados a largo plazo» relativos a la liberación y a la comercialización. Por «efectos acumulados a largo plazo» se entienden los efectos acumulados que las autorizaciones puedan tener en la salud humana y el medio ambiente, incluidos, entre otros elementos, la flora y la fauna, la fertilidad del suelo, la capacidad del suelo para degradar materias orgánicas, la cadena alimentaria tanto para los animales como para el ser humano, la diversidad biológica, la salud animal y los problemas de resistencia a los antibióticos.
A. Objetivo
El objetivo de una evaluación del riesgo es, caso por caso, identificar y evaluar efectos adversos potenciales del organismo modificado genéticamente, ya sean directos o indirectos, inmediatos o diferidos, en la salud humana y el medio ambiente que la liberación voluntaria o la comercialización de organismos modificados genéticamente puede tener.
La evaluación del riesgo deberá llevarse a cabo con objeto de identificar si hay una necesidad de gestión del riesgo y, en caso afirmativo, los métodos más apropiados que deben utilizarse.
B. Principios generales
De acuerdo con el principio de cautela, al llevar a cabo la evaluación del riesgo, deben seguirse los siguientes principios generales:
1.° Las características identificadas del organismo modificado genéticamente y su uso que tengan un potencial de efectos adversos deberán compararse a los que presente el organismo no modificado del cual se deriva y su uso en situaciones similares.
2.° La evaluación del riesgo deberá llevarse a cabo en condiciones de seguridad y transparencia científica, basándose en los datos científicos y técnicos disponibles.
3.° La evaluación del riesgo deberá llevarse a cabo caso por caso, de forma que la información requerida pueda variar en función del tipo de los organismos modificados genéticamente de que se trate, de su uso previsto y del medio ambiente de recepción potencial, teniendo en cuenta, entre otras cosas, los organismos modificados genéticamente que ya se encuentren en el medio ambiente.
4.° En caso de disponer de nueva información sobre el organismo modificado genéticamente y sus efectos en la salud humana o el medio ambiente, puede que sea necesario realizar una nueva evaluación del riesgo para:
Determinar si el riesgo ha cambiado.
Determinar si es necesario modificar en consecuencia la gestión del riesgo.
C. Metodología
Están disponibles unas directrices publicadas por la Autoridad Europea de Seguridad Alimentaria para la aplicación de la presente sección para las notificaciones previstas en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, descrita en el capítulo II del título II del presente reglamento.
C.1 Consideraciones generales y específicas para la evaluación de riesgo.
1. Cambios intencionados y no intencionados.
Como parte de la identificación y evaluación de los posibles efectos adversos a los que se hace referencia en la sección A, la evaluación de riesgo identificará los cambios intencionados y no intencionados resultantes de la modificación genética y evaluará su potencial para causar efectos adversos sobre la salud humana, la sanidad animal y el medio ambiente.
Los cambios intencionados resultantes de la modificación genética son aquéllos diseñados para que ocurran y que cumplen los objetivos originales de la modificación genética.
Los cambios no intencionados resultantes de la modificación genética son aquellos cambios que van más allá de los cambios intencionados causados por la modificación genética.
Los cambios intencionados y no intencionados pueden tener efectos directos o indirectos y efectos inmediatos o diferidos en la salud humana, la sanidad animal y el medio ambiente.
2. Efectos adversos a largo plazo y efectos adversos acumulados a largo plazo en la evaluación de riesgo de las notificaciones previstas en la parte C de la Directiva 2001/18/CE, del Parlamento Europeo y del Consejo, de 12 de marzo de 2001.
Los efectos a largo plazo de un OMG son efectos debidos bien a una respuesta diferida de los organismos o su progenie a una exposición crónica o a largo plazo al OMG, o bien de un amplio uso del OMG en el tiempo y el espacio.
En la identificación y evaluación de los posibles efectos adversos a largo plazo de un OMG en la salud humana, la sanidad animal y el medio ambiente se tendrán en cuenta los siguientes aspectos:
a) Las interacciones a largo plazo entre los OMG y el entorno receptor;
b) Las características del OMG que adquieran importancia a largo plazo;
c) Los datos obtenidos de las comercializaciones o liberaciones intencionales repetidas del OMG durante un largo período de tiempo.
En la identificación y evaluación de los posibles efectos adversos acumulados a largo plazo a que hace referencia la parte introductoria de este anexo, se tendrán asimismo en cuenta las comercializaciones o liberaciones intencionales de OMG en el pasado.
3. Calidad de los datos.
Con objeto de llevar a cabo una evaluación de riesgo en relación con una notificación prevista en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, el notificador deberá recabar la información que ya se encuentra disponible en la documentación científica u otras fuentes, incluidos los informes de seguimiento, y generará los datos necesarios mediante la realización, siempre que sea posible, de los estudios adecuados. En su caso, el notificador deberá justificar en la evaluación de riesgo la imposibilidad de generar datos a partir de estudios.
La evaluación de riesgo correspondiente a las notificaciones en virtud de la parte B de la presente Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, descrita en el capítulo II, del título II del presente reglamento se basará, al menos, en datos ya disponibles en la documentación científica u otras fuentes y se podrá completar con datos adicionales generados por el notificador.
En caso de que la evaluación de riesgo incluya datos generados fuera de Europa, deberá justificarse su pertinencia para el entorno o los entornos receptores en la Unión.
Los datos incluidos en la evaluación de riesgo correspondiente a las notificaciones previstas en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, deberán cumplir los siguientes requisitos:
a) Siempre que la evaluación de riesgo incluya estudios toxicológicos efectuados con objeto de evaluar el riesgo para la salud humana o de los animales, el notificador deberá presentar pruebas que acrediten que éstos se realizaron en instalaciones conformes a:
i) Los requisitos de la Directiva 2004/10/CE del Parlamento Europeo y del Consejo, de 11 de febrero de 2004, relativa a la inspección y verificación de las buenas prácticas de laboratorio (BPL) o
ii) Los “principios de la OCDE sobre buenas prácticas de laboratorio” (BPL), si los estudios se realizaron fuera de la Unión;
b) Si la evaluación de riesgo incluye estudios distintos de los toxicológicos, estos deberán:
i) En su caso, cumplir los principios relativos a las buenas prácticas de laboratorio (BPL) establecidos en la Directiva 2004/10/CE del Parlamento Europeo y del Consejo, de 11 de febrero de 2004, o
ii) Ser efectuados por organismos acreditados con arreglo a la norma ISO pertinente, o
iii) En ausencia de una norma ISO pertinente, ser efectuados de conformidad con normas reconocidas internacionalmente;
c) La información sobre los resultados obtenidos en los estudios mencionados en las letras a) y b) y sobre los protocolos de estudio utilizados será fiables y exhaustivos e incluirán los datos brutos en un formato electrónico adecuado para realizar un análisis estadístico o de otro tipo;
d) El notificador especificará, en la medida de lo posible, la magnitud del efecto que se prevé detectar con cada estudio realizado, y lo justificará.
e) La selección de emplazamientos para los estudios de campo deberá basarse en entornos receptores pertinentes, teniendo en cuenta la exposición y el impacto potenciales observables en el lugar en el que pueda liberarse el OMG. La selección deberá justificarse en la evaluación de riesgo;
f) El referente de comparación no modificado genéticamente deberá ser adecuado para el entorno receptor pertinente y tener unos antecedentes genéticos comparables a los del OMG. La selección del referente de comparación deberá justificarse en la evaluación de riesgo;
4. Eventos de transformación apilados en las notificaciones previstas en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001.
Los siguientes requisitos se aplicarán a las evaluaciones de riesgo de un OMG que contengan eventos de transformación apilados en las notificaciones previstas en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y el Consejo, de 12 de marzo de 2001:
a) El notificador proporcionará una evaluación de riesgo para cada evento de transformación único del OMG o hará referencia a las notificaciones ya presentadas de esos eventos de transformación únicos;
b) El notificador suministrará una evaluación de los siguientes aspectos:
i) La estabilidad de los eventos de transformación,
ii) La expresión de los eventos de transformación,
iii) Los posibles efectos aditivos, sinérgicos o antagonistas resultantes de la combinación de los eventos de transformación;
c) Si la progenie del OMG puede contener varias subcombinaciones de los eventos de transformación apilados, el notificador aportará una justificación científica de que no es necesario aportar datos experimentales en relación con las subcombinaciones de que se trate, independientemente de su origen; a falta de tal justificación científica, proporcionará los datos experimentales pertinentes.
C.2 Características de los OMG y de las liberaciones.
La evaluación de riesgo deberá tener en cuenta los detalles técnicos y científicos pertinentes que guarden relación con las características de:
– el organismo o los organismos receptores o parentales,
– la modificación o las modificaciones genéticas, ya sea por inserción o por supresión de material genético, y la información pertinente sobre el vector y el donante,
– el OMG en cuestión,
– el uso o la liberación intencional, inclusive su escala,
– el posible o los posibles entornos receptores en los que se vaya a liberar el OMG y el entorno en el que pueda propagarse el transgén, así como
– las interacciones entre estas características.
Sin perjuicio de lo dispuesto en el artículo 23.3 y el artículo 32.2.g), la evaluación de riesgo incluirá la información pertinente relativa a liberaciones previas del mismo OMG o los mismos OMG, o de un OMG o de OMG similares, y de organismos con características parecidas y su interacción biótica y abiótica con entornos receptores similares, incluida la información resultante del seguimiento de dichos organismos.
C.3 Fases de la evaluación del riesgo.
La evaluación de riesgo contemplada en los artículos 22, 23 y 32, se realizará para cada ámbito de riesgo pertinente a que se hace referencia en las secciones D1 o D2 con arreglo a las seis fases siguientes:
1. Definición de problemas, incluida la identificación de peligros.
La definición del problema incluirá:
a) La identificación de cualquier cambio en las características del organismo que estén relacionadas con la modificación genética, mediante la comparación de las características del OMG con las del organismo no modificado genéticamente elegido como referente, en condiciones similares de liberación o uso;
b) La identificación de los posibles efectos adversos en la salud humana, la sanidad animal o el medio ambiente, vinculados a los cambios que se hayan identificado con arreglo a la letra a);
Los posibles efectos adversos no se deben descartar sobre la base de que es improbable que ocurran.
Los posibles efectos adversos variarán en cada caso y pueden incluir:
– efectos en la dinámica de poblaciones de especies en el entorno receptor y en la diversidad genética de cada una de esas poblaciones, que conducen a un posible descenso de la biodiversidad,
– susceptibilidad alterada respecto a patógenos que faciliten la difusión de enfermedades infecciosas o que creen nuevos reservorios o vectores,
– disminución de la eficacia de tratamientos médicos, veterinarios o de protección fitosanitaria, profilácticos o terapéuticos, por ejemplo, mediante la transferencia de genes que confieran resistencia a los antibióticos utilizados en la medicina humana o veterinaria,
– efectos en biogeoquímica (ciclos biogeoquímicos), incluido el reciclado del carbón y del nitrógeno mediante cambios en la descomposición del material orgánico del suelo,
– enfermedades que afecten a los seres humanos, incluidas las reacciones alergénicas o tóxicas,
– enfermedades que afecten a los animales y a las plantas, incluidos los efectos tóxicos, y en el caso de los animales, las reacciones alergénicas, en su caso.
En caso de que se identifiquen posibles efectos adversos a largo plazo de un OMG, estos deberán evaluarse mediante estudios documentales que tengan en cuenta, en la medida de lo posible, al menos una de las siguientes fuentes:
i) pruebas procedentes de experiencias previas,
ii) documentación o conjuntos de datos disponibles;
iii) modelización matemática;
c) La identificación de los parámetros de evaluación pertinentes.
Los posibles efectos adversos que podrían repercutir en los parámetros de evaluación identificados se tendrán en cuenta en las próximas etapas de la evaluación de riesgo;
d) La identificación y descripción de las vías de exposición u otros mecanismos que pueden hacer que ocurran efectos adversos.
Los efectos adversos pueden ocurrir directa o indirectamente a través de vías de exposición u otros mecanismos que pueden incluir:
– la propagación del o de los OMG en el medio ambiente,
– la transferencia del material genético insertado al mismo organismo o a otros organismos, estén o no modificados genéticamente,
– inestabilidad fenotípica o genética,
– interacciones con otros organismos,
– cambios en la gestión y también, en su caso, en las prácticas agrícolas;
e) La formulación de hipótesis comprobables y la definición de parámetros de medición pertinentes para permitir, en la medida de lo posible, una evaluación cuantitativa de los posibles efectos adversos;
f) La consideración de posibles incertidumbres, incluidas las lagunas de conocimiento y las limitaciones metodológicas.
2. Caracterización del peligro.
Deberá evaluarse la magnitud de cada posible efecto adverso. Dicha evaluación deberá presuponer que dicho efecto adverso se va a producir. La evaluación de riesgo deberá considerar que es probable que la magnitud esté influenciada por el entorno receptor en el que está previsto que se libere el OMG y por la escala y las condiciones de la liberación.
En la medida de lo posible, la evaluación se expresará en términos cuantitativos.
Si la evaluación se expresa en términos cualitativos, deberá utilizarse una descripción categórica (“alta”, “moderada”, “baja” o “insignificante”) y se suministrará una explicación de la magnitud de los efectos que representa cada categoría.
3. Caracterización de la exposición.
Se evaluará la posibilidad o probabilidad de que se produzca cada posible efecto adverso identificado, a fin de proporcionar, en su caso, una evaluación cuantitativa de la exposición como una medida de probabilidad relativa, o bien una evaluación cualitativa de la exposición. Se tendrán en cuenta las características del entorno receptor y el ámbito de la notificación.
Si la evaluación se expresa en términos cualitativos, deberá utilizarse una descripción categórica (“alta”, “moderada”, “baja” o “insignificante”) de la exposición y se suministrará una explicación de la magnitud del efecto que representa cada categoría.
4. Caracterización del riesgo.
El riesgo se caracterizará combinando, para cada posible efecto adverso, la magnitud y la probabilidad de que se produzca este efecto adverso con objeto de suministrar una estimación cuantitativa, o semicuantitativa, del riesgo.
En caso de que no se pueda hacer una estimación cuantitativa ni semicuantitativa, se presentará una estimación cualitativa del riesgo. En este caso, deberá utilizarse una descripción categórica del riesgo (“alto”, “moderado”, “bajo” o “insignificante”) y se suministrará una explicación de la magnitud de los efectos que representa cada categoría.
En su caso, se describirá la incertidumbre de cada riesgo identificado y, en la medida de lo posible, se expresará en términos cuantitativos.
5. Estrategias de gestión de riesgos.
En caso de que se identifiquen riesgos que requieran, sobre la base de su caracterización, medidas para gestionarlos, se propondrá una estrategia de gestión de riesgos.
Las estrategias de gestión del riesgo se describirán en términos de reducción del peligro o de la exposición, o ambos, y serán proporcionadas a la reducción del riesgo, la escala y las condiciones de la liberación y los niveles de incertidumbre identificados en la evaluación de riesgo.
La consiguiente reducción del riesgo global se cuantificará siempre que sea posible.
6. Evaluación global del riesgo y conclusiones.
Se realizará una evaluación cualitativa y, siempre que sea posible, cuantitativa, del riesgo global del OMG teniendo en cuenta los resultados de la caracterización del riesgo, las estrategias de gestión del riesgo propuestas y los niveles de incertidumbre asociados.
La evaluación global del riesgo incluirá, en su caso, las estrategias de gestión del riesgo propuestas para cada riesgo identificado.
En la evaluación global del riesgo y las conclusiones se propondrán asimismo requisitos específicos para el plan de seguimiento del OMG, y, en su caso, para el seguimiento de la eficacia de las medidas de gestión del riesgo propuestas.
Por lo que se refiere a las notificaciones previstas en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, la evaluación global del riesgo incluirá asimismo una explicación de las hipótesis formuladas durante la evaluación de riesgo, así como de la naturaleza y la magnitud de las incertidumbres asociadas con los riesgos, y una justificación de las medidas de gestión de riesgos propuestas.
D. Conclusiones relativas a los ámbitos específicos de riesgo de la Evaluación del riesgo.
Se extraerán conclusiones relativas al impacto potencial sobre el medio ambiente, en los entornos receptores pertinentes, de la liberación o la comercialización de OMG en relación con cada uno de los ámbitos de riesgo pertinentes enumerados en la sección D1 para los OMG distintos de las plantas superiores, o en la sección D2 para las plantas superiores modificadas genéticamente, sobre la base de una evaluación de riesgo efectuada de conformidad con los principios establecidos en la sección B, aplicando la metodología descrita en la sección C y sobre la base de la información necesaria en virtud del anexo V.
D.1. En el caso de organismos modificados genéticamente diferentes de las plantas superiores
1. Probabilidad de que el organismo modificado genéticamente se convierta en persistente e invasor en hábitats naturales en las condiciones de liberación(es) propuesta(s).
2. Cualquier ventaja o desventaja selectiva que haya adquirido el organismo modificado genéticamente y la probabilidad de que se convierta en realidad en las condiciones de liberación(es) propuesta(s).
3. Transferencia potencial de genes a otras especies en las condiciones de liberación propuestas del organismo modificado genéticamente y cualquier ventaja o desventaja selectiva que adquieran dichas especies.
4. Impacto potencial en el medio ambiente inmediato y/o diferido de las interacciones directas e indirectas entre el organismo modificado genéticamente y los organismos objeto de la investigación si procede.
5. Impacto potencial sobre el medio ambiente inmediato y/o diferido de las interacciones directas e indirectas entre el organismo modificado genéticamente y los organismos ajenos a la investigación, incluido el impacto en los niveles de población de los competidores, presas, huéspedes, simbiontes, predadores, parásitos y organismos patógenos.
6. Posibles efectos inmediatos y/o diferidos sobre la salud humana a consecuencia de las interacciones potenciales directas e indirectas entre los organismos modificados genéticamente y las personas que trabajan con ellos, están en contacto con ellos o cerca de la(s) liberación(es) de organismos modificados genéticamente.
7. Posibles efectos inmediatos y/o diferidos para la salud animal y consecuencias sobre la cadena alimentaria humana o animal resultado del consumo del organismo modificado genéticamente y de cualquier producto derivado de él que se prevea utilizar como alimento animal.
8. Posibles efectos inmediatos y/o diferidos sobre los procesos biogeoquímicos resultado de las potenciales interacciones directas e indirectas entre el organismo modificado genéticamente y organismos objeto de la investigación o ajenos a ella cerca de la o de las liberación(es) de organismos modificados genéticamente.
9. Posible impacto directo e indirecto sobre el medio ambiente inmediato y/o diferido de las técnicas especificas utilizadas para la gestión del organismo modificado genéticamente cuando sean diferentes de las empleadas para los organismos que no hayan sido modificados genéticamente.
D.2. En el caso de las plantas superiores modificadas genéticamente (PSMG).
La expresión "plantas superiores" se refiere a las plantas pertenecientes al grupo taxonómico de los espermatofitos (gimnospermas y angiospermas).
1. Persistencia y capacidad de invasión de la PSMG, incluida la transferencia de información genética entre plantas.
2. Transferencia genética de las plantas a los microorganismos.
3. Interacciones de las PSMG con los organismos objetivo.
4. Interacciones de las PSMG con los organismos no objetivo.
5. Impacto de las técnicas específicas de cultivo, gestión y recolección.
6. Efectos sobre los procesos biogeoquímicos.
7. Efectos en la salud humana y animal.
Se modifican los apartados C y D por el art. 2.15 y 16 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Esta modificación entra en vigor el 30 de septiembre de 2019, según establece la disposición final 2 del citado Real Decreto.
[Bloque 97: #anv]
Las solicitudes de autorización de la parte B y C de la Directiva 2001/18/CE, del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, incluirán, por regla general, la información prevista en el anexo V.A, en el caso de OMG distintos de plantas superiores o en el anexo V.B, en el caso de plantas superiores modificadas genéticamente.
No será necesario presentar un determinado subconjunto de los datos enumerados en el anexo V.A o en el anexo V.B, siempre que éste no sea pertinente ni necesario a efectos de una evaluación del riesgo en el marco de una determinada notificación, habida cuenta en particular de las características del OMG, de la escala y las condiciones de la liberación o de sus condiciones de uso previstas.
El nivel de precisión adecuado para cada subconjunto de datos también podrá variar según la naturaleza y la escala de la liberación propuesta.
Para cada subconjunto de datos requerido se suministrará la información que figura a continuación:
i) los resúmenes y resultados de los estudios mencionados en la notificación, incluida una explicación sobre su pertinencia para la evaluación de riesgo, en su caso,
ii) en relación con las notificaciones a que se hace referencia en la parte C de la Directiva 2001/18/CE del Parlamento Europeo y del Consejo, de 12 de marzo de 2001, anexos con información detallada sobre estos estudios, incluida la descripción de los métodos y los materiales utilizados o la referencia a métodos normalizados o reconocidos internacionalmente, así como el nombre de la entidad o entidades responsables de realizar los estudios.
Los futuros acontecimientos en el ámbito de la modificación genética podrán hacer necesario adaptar el presente anexo a los avances técnicos o elaborar notas orientativas sobre este. Una vez adquirida suficiente experiencia en la Unión sobre las notificaciones de liberación de OMG concretos, se podrá introducir una mayor precisión respecto de los requisitos de información necesarios para diferentes tipos de OMG; por ejemplo, árboles y plantas perennes, organismos unicelulares, peces o insectos, o en relación con un uso particular de los OMG, como el desarrollo de vacunas.
Se modifica por el art. 2.17 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Esta modificación entra en vigor el 30 de septiembre de 2019, según establece la disposición final 2 del citado Real Decreto.
[Bloque 98: #anva]
I. INFORMACIÓN DE CARÁCTER GENERAL
A. Nombre y dirección del interesado (empresa o institución).
B. Nombre, titulación y experiencia del científico o científicos responsables.
C. Título del proyecto.
II. INFORMACIÓN RELATIVA A LOS ORGANISMOS MODIFICADOS GENÉTICAMENTE
A. Características del organismo o de los organismos: a) donantes; b) receptores, o c) (cuando proceda) parentales:
1. Nombre científico.
2. Taxonomía.
3. Otros nombres (nombre común, nombre de la cepa, etc.).
4. Características fenotípicas y genéticas.
5. Grado de parentesco entre los organismos donantes y receptores o entre los organismos parentales.
6. Descripción de las técnicas de identificación y detección.
7. Sensibilidad, fiabilidad (en términos cuantitativos) y especificidad de las técnicas de detección e identificación.
8. Descripción de la distribución geográfica y del hábitat natural del organismo, incluida información sobre depredadores naturales, presas, parásitos, competidores, simbiontes y huéspedes.
9. Organismos con los que se sabe que la transferencia de material genético se da en condiciones naturales.
10. Verificación de la estabilidad genética de los organismos y factores que influyen en ella.
11. Rasgos patológicos, ecológicos y fisiológicos de los organismos:
a) Clasificación de los riesgos, de conformidad con las normas comunitarias vigentes relativas a la protección de la salud humana y/o el medio ambiente.
b) Período de generación en ecosistemas naturales, ciclo reproductivo sexual y asexual.
c) Información sobre la supervivencia, incluidas la estacionalidad y la capacidad para formar estructuras de supervivencia.
d) Patogenicidad: infectividad, toxigenicidad, virulencia, alergenicidad, portador (vector) de patógeno, vectores posibles, gama de huéspedes incluidos los organismos que no sean objeto de la investigación.
Posible activación de virus latentes (provirus). Capacidad para colonizar otros organismos.
e) Resistencia a los antibióticos y uso potencial de dichos antibióticos en seres humanos y organismos domésticos con fines profilácticos y terapéuticos.
f) Participación en procesos ambientales: producción primaria, ciclos de nutrientes, descomposición de la materia orgánica, respiración, etc.
12. Naturaleza de los vectores indígenas:
a) Secuencia.
b) Frecuencia de movilización.
c) Especificidad.
d) Presencia de genes que confieren resistencia.
13. Historial de modificaciones genéticas anteriores.
B. Características del vector:
1. Naturaleza y procedencia del vector.
2. Secuencia de transposones, vectores y demás fragmentos genéticos no codificadores empleados para producir los organismos modificados genéticamente y para hacer funcionar en ellos el vector y el fragmento de inserción introducidos.
3. Frecuencia de movilización del vector insertado y/o capacidad de transmisión genética, así como los métodos para su determinación.
4. Información sobre el grado en que el vector está limitado al ADN necesario para realizar la función deseada.
C. Características del organismo modificado:
1. Información relativa a la modificación genética:
a) Métodos de modificación empleados.
b) Métodos empleados para preparar y efectuar la inserción o inserciones en el receptor o para borrar una secuencia.
c) Descripción de la preparación del fragmento de inserción y/o del vector.
d) Ausencia en el fragmento de inserción de toda secuencia desconocida, e información acerca del grado en que la secuencia insertada se limita al ADN necesario para llevar a cabo la función deseada.
e) Métodos y criterios utilizados en la selección.
f) Secuencia, identidad funcional y localización del segmento o segmentos de ácido nucleico alterados, insertados o borrados de que se trate, con especial referencia a cualquier secuencia nociva conocida.
2. Información sobre el organismo modificado genéticamente final:
a) Descripción de los rasgos genéticos o características fenotípicas y en especial de todos aquellos rasgos y características nuevas que puedan expresarse o los que ya no puedan ser expresados.
b) Estructura y cantidad de todo vector y/o ácido nucleico donante que quede en la composición final del organismo modificado.
c) Estabilidad del organismo desde el punto de vista de los rasgos genéticos.
d) Coeficiente y nivel de expresión del nuevo material genético. Métodos y sensibilidad de medición.
e) Actividad de la proteína o proteínas expresadas.
f) Descripción de las técnicas de identificación y detección, incluidas las técnicas de identificación y detección de la secuencia y del vector insertados.
g) Sensibilidad, fiabilidad (en términos cuantitativos) y especificidad de las técnicas de identificación y detección.
h) Historial de las liberaciones o usos anteriores del organismo modificado genéticamente.
i) Aspectos relativos a la salud humana y la salud animal, así como aspectos fitosanitarios:
1.° Efectos alergénicos o tóxicos de los organismos modificados genéticamente y/o sus productos metabólicos.
2.° Comparación de la patogenicidad del organismo modificado con la del organismo donante, receptor o (si procede) parental.
3.° Capacidad de colonización.
4.° En caso de que el organismo sea patógeno para personas inmunocompetentes: enfermedades causadas y mecanismos patogénicos, incluidas la capacidad de invasión y la virulencia,
gapacidad de comunicación,
dosis infecciosa,
gama de huéspedes, posibilidad de alteración,
posibilidad de supervivencia fuera del huésped humano,
presencia de vectores o medios de diseminación,
estabilidad biológica,
patrones de resistencia a los antibióticos,
alergenicidad,
existencia de terapias apropiadas.
5.° Otros peligros resultantes del producto.
III. INFORMACIÓN RELATIVA A LAS CONDICIONES DE LIBERACIÓN VOLUNTARIA Y AL MEDIO AMBIENTE RECEPTOR
A. Información sobre la liberación voluntaria:
1. Descripción de la liberación voluntaria propuesta, incluido el fin o los fines y los productos previstos.
2. Fechas previstas de la liberación y calendario del experimento, con la frecuencia y la duración de las liberaciones.
3. Preparación del lugar antes de la liberación.
4. Extensión del lugar.
5. Métodos que vayan a emplearse para la liberación.
6. Cantidades de organismos modificados genéticamente que vayan a ser liberadas.
7. Alteraciones causadas en el lugar (tipo y método de cultivo, minería, irrigación u otras actividades).
8. Medidas de protección de los operarios durante la liberación.
9. Tratamiento del lugar después de la liberación.
10. Técnicas previstas para la eliminación o la desactivación de los organismos modificados genéticamente tras el experimento.
11. Información y resultados de anteriores liberaciones de los organismos modificados genéticamente, sobre todo en distintas escalas y en ecosistemas diferentes.
B. Información sobre el medio ambiente (tanto in situ como en un entorno más amplio):
1. Ubicación geográfica y coordenadas de referencia del lugar o lugares (en el caso de las solicitudes con arreglo al capítulo III, se considerarán lugares de liberación las zonas donde esté previsto el uso del producto).
2. Proximidad fisica o biológica a seres humanos y flora y fauna importantes.
3. Proximidad de biotopos, zonas protegidas o suministros de agua potable importantes.
4. Características climáticas de la región o regiones que podrían verse afectadas.
5. Características geográficas, geológicas y edafológicas.
6. Flora y fauna, incluidas cosechas, ganado y especies migratorias.
7. Descripción de los ecosistemas que podrían verse afectados, tanto si son objeto de la investigación como si no lo son.
8. Comparación del hábitat natural del organismo receptor con el lugar o lugares propuestos para la liberación.
9. Cualquier proyecto urbanístico o de modificación del empleo del suelo de la región que pudiera tener influencia en el efecto ambiental de la liberación.
IV. INFORMACIÓN RELATIVA A LA INTERACCIÓN ENTRE LOS ORGANISMOS MODIFICADOS GENÉTICAMENTE Y EL MEDIO AMBIENTE
A. Características que afecten a la supervivencia, a la multiplicación y a la diseminación:
1. Características biológicas que afecten a la supervivencia, a la multiplicación y a la dispersión.
2. Condiciones ambientales conocidas o previstas que puedan afectar a la supervivencia, a la multiplicación y a la diseminación (viento, agua, suelo, temperatura, pH, etc.).
3. Sensibilidad a agentes específicos.
B. Interacciones con el medio ambiente:
1. Hábitat previsto de los organismos modificados genéticamente.
2. Estudios sobre el comportamiento y características de los organismos modificados genéticamente y sobre su impacto ecológico, llevados a cabo en ambientes naturales simulados, tales como microcosmos, cámaras de crecimiento, invernaderos, etc.
3. Capacidad de transmisión genética:
a) Transmisión de material genético de los organismos modificados genéticamente a los organismos de los ecosistemas afectados, con posterioridad a la liberación.
b) Transmisión de material genético de los organismos propios del ecosistema a los organismos modificados genéticamente, con posterioridad a la liberación.
4. Probabilidad de que después de la liberación se produzca una selección que se manifieste en la expresión de rasgos inesperados y/o indeseables en el organismo modificado.
5. Medidas utilizadas para garantizar y verificar la estabilidad genética. Descripción de los rasgos genéticos que puedan impedir o reducir al mínimo la dispersión del material genético. Métodos de verificación de la estabilidad genética.
6. Rutas de dispersión biológica, modelos conocidos o posibles de interacción con el agente de diseminación: entre ellos la inhalación, la ingestión, el contacto superficial, la penetración a través de la piel, etc.
7. Descripción de los ecosistemas en los que podrían ser diseminados los organismos modificados genéticamente.
8. Posibilidad de un incremento excesivo de la población en el medio ambiente.
9. Ventaja competitiva de los organismos modificados genéticamente en relación con el organismo u organismos receptores o parentales no modificados.
10. Identificación y descripción de los organismos objeto de la investigación, si procede.
11. Mecanismo previsto y resultado de la interacción entre los organismos modificados genéticamente liberados y el organismo u organismos objeto de la investigación, si procede.
12. Identificación y descripción de los organismos que no sean objeto de la investigación y puedan verse afectados negativamente por la liberación de los organismos modificados genéticamente y de los mecanismos previstos de la interacción negativa que se identifiquen.
13. Posibilidades de cambios posteriores a la liberación en las interacciones biológicas o en la gama de los huéspedes.
14. Interacciones conocidas o previstas con organismos del medio ambiente que no sean objeto de la investigación como, por ejemplo, competidores, presas, huéspedes, simbiontes, predadores, parásitos y agentes patógenos.
15. Implicaciones conocidas o previstas en procesos biogeoquímicos.
16. Otras posibles interacciones con el medio ambiente.
V. INFORMACIÓN SOBRE SEGUIMIENTO, CONTROL, TRATAMIENTO DE RESIDUOS Y PLANES DE ACCIÓN DE EMERGENCIA
A. Técnicas de control:
1. Métodos de rastreo de los organismos modificados genéticamente y de seguimiento de sus efectos.
2. Especificidad (para identificar a los organismos modificados genéticamente y para distinguirlos del organismo donante, receptor o, si procede, parental), sensibilidad y fiabilidad de las técnicas de control.
3. Técnicas de detección de la transmisión a otros organismos del material genético donado.
4. Duración y frecuencia del control.
B. Control de la liberación:
1. Métodos y procedimientos para evitar y/o reducir al mínimo la diseminación de los organismos modificados genéticamente fuera del lugar de la liberación o de la zona prevista para su uso.
2. Métodos y procedimientos para proteger el lugar mencionado contra la entrada de personas no autorizadas.
3. Métodos y procedimientos para impedir que otros organismos penetren en dicho lugar.
C. Tratamiento de residuos:
1. Tipo de residuos producidos.
2. Volumen de residuos previsto.
3. Descripción del tratamiento propuesto.
D. Planes de acción en caso de emergencia:
1. Métodos y procedimientos de control de los organismos modificados genéticamente en caso de diseminación inesperada.
2. Métodos de descontaminación de las zonas afectadas, por ejemplo, erradicación de los organismos modificados genéticamente.
3. Métodos de eliminación o de saneamiento de plantas, animales, suelos, etc., expuestos al organismo durante la diseminación o después de la misma.
4. Métodos de aislamiento de la zona afectada por la diseminación.
5. Planes de protección de la salud humana y del medio ambiente en caso de que se produzca un efecto indeseable.
[Bloque 99: #anvb]
I. Información requerida en las notificaciones presentadas con arreglo a los artículos 23 y 28
A. Información general
1. Nombre y dirección del notificador (empresa o institución).
2. Nombre, titulación y experiencia del científico o científicos responsables.
3. Título del proyecto.
4. Información relativa a la liberación.
a) finalidad de la liberación;
b) fecha o fechas y duración previstas de la liberación;
c) método mediante el cual se liberarán las PSMG;
d) método de preparación y gestión del lugar de liberación, con carácter previo, simultáneo y posterior a la liberación, con inclusión de prácticas de cultivo y métodos de recolección;
e) número aproximado de plantas (o plantas por m2).
5. Información relativa al lugar de la liberación
a) localización y extensión del lugar o lugares de liberación;
b) descripción del ecosistema del lugar de liberación, con inclusión de datos sobre el clima, la flora y la fauna;
c) presencia de especies vegetales, tanto silvestres emparentadas como especies cultivadas, compatibles sexualmente;
d) proximidad de biotopos reconocidos oficialmente o de zonas protegidas que puedan verse afectados.
B. Información científica
1. Información relativa a la planta receptora o (en su caso), a las plantas parentales.
a) Nombre completo:
i) nombre de la familia,
ii) género,
iii) especie,
iv) subespecies,
v) cultivar o línea de reproducción,
vi) nombre vulgar.
b) Distribución geográfica y cultivo de la planta en la Unión.
c) Información sobre la reproducción:
i) modo o modos de reproducción,
ii) de haberlos, factores específicos que afecten a la reproducción,
iii) período de generación.
d) Compatibilidad sexual con otras especies vegetales cultivadas o silvestres, indicando la distribución de las especies compatibles en Europa.
e) Capacidad de supervivencia:
i) posibilidad de formar estructuras de supervivencia o de latencia,
ii) factores específicos, en su caso, que afecten a la supervivencia.
f) Diseminación:
i) formas y alcance de la diseminación,
ii) de haberlos, los factores específicos que afecten a la diseminación.
g) En el caso de especies vegetales que no crezcan normalmente en la Unión, una descripción del hábitat natural de la planta que incluya información sobre predadores naturales, parásitos, competidores y simbiontes.
h) Posibles interacciones de la planta, pertinentes para la PSMG, con organismos presentes en el ecosistema en el que crece normalmente, o en otros lugares, incluida la información sobre los efectos tóxicos para las personas, los animales y otros organismos.
2. Caracterización molecular.
a) Información relativa a la modificación genética:
i) descripción de los métodos utilizados para la modificación genética,
ii) naturaleza y origen del vector utilizado,
iii) origen del ácido nucleico o los ácidos nucleicos utilizados para la transformación, tamaño y función prevista de cada fragmento componente de la región que se pretenda insertar.
b) Información relativa a la PSMG.
i) descripción general del rasgo o los rasgos y las características que se hayan introducido o modificado,
ii) información sobre las secuencias insertadas/suprimidas realmente:
– el tamaño y el número de copias de todos los insertos y los métodos utilizados para su caracterización,
– en caso de supresión, tamaño y función de la región o regiones suprimidas,
– localización subcelular del inserto o los insertos en las células vegetales (integrados en el núcleo, cloroplastos o mitocondrias, o mantenidos en forma no integrada) y métodos para su determinación,
iii) partes de la planta en que se expresa el inserto,
iv) estabilidad genética del fragmento de inserción y estabilidad fenotípica de la PSMG.
c) Conclusiones de la caracterización molecular.
3. Información relativa a ámbitos de riesgo específicos:
a) Cualquier cambio en la persistencia o la capacidad de invasión de la PSMG, así como su habilidad para transferir material genético a variedades emparentadas compatibles sexualmente y los efectos medioambientales adversos que se derivan.
b) Cualquier cambio en la capacidad de la PSMG de transferir material genético a microorganismos y los efectos medioambientales adversos que se derivan.
c) El mecanismo de interacción entre la PSMG y los organismos objeto de la investigación (en su caso) y los efectos medioambientales adversos que se derivan.
d) Posibles cambios en las interacciones de la PSMG con organismos no objetivo debidos a la modificación genética y los efectos medioambientales adversos que se derivan.
e) Posibles cambios en las prácticas agrícolas y la gestión de las PSMG debidos a la modificación genética y los efectos medioambientales adversos que se derivan.
f) Posibles interacciones con el entorno abiótico y los efectos medioambientales adversos que se derivan.
g) Información sobre cualquier posible efecto tóxico o alergénico u otros efectos adversos para la salud humana y de los animales derivado de la modificación genética.
h) Conclusiones relativas a los ámbitos de riesgo específicos.
4. Información sobre los planes para el control, el seguimiento y el tratamiento de residuos, así como para después de la liberación.
a) Cualquier medida adoptada, con inclusión de:
i) el aislamiento espacial y temporal de las especies vegetales sexualmente compatibles, tanto malas hierbas y variedades silvestres emparentadas como variedades cultivadas,
ii) cualquier medida para minimizar o prevenir la dispersión de todo material de reproducción de las PSMG.
b) Descripción de los métodos para el tratamiento del lugar después de la liberación.
c) Descripción de los métodos de tratamiento del material vegetal modificado genéticamente después de la liberación, incluidos los residuos.
d) Descripción de los planes y técnicas de seguimiento.
e) Descripción de los planes de emergencia.
f) Descripción de los métodos y procedimientos para:
i) evitar o reducir al mínimo la diseminación de las PSMG más allá del lugar de liberación,
ii) proteger el lugar de la entrada de personas no autorizadas,
iii) impedir que otros organismos penetren en dicho lugar o minimizar tales entradas.
5. Descripción de las técnicas de detección e identificación de las PSMG.
6. Información sobre liberaciones previas de la PSMG, en su caso.
II. Información requerida en las notificaciones presentadas con arreglo a lo dispuesto en el artículo 32
A. Información general
1. Nombre y dirección del notificador (empresa o institución).
2. Nombre, titulación y experiencia del científico o científicos responsables.
3. Designación y especificación de la PSMG.
4. Ámbito de la notificación:
a) cultivo,
b) otros usos (que deberán especificarse en la notificación).
B. Información científica
1. Información relativa a la planta receptora o (en su caso), a las plantas parentales.
a) nombre completo:
i) nombre de la familia,
ii) género,
iii) especie,
iv) subespecie,
v) cultivar/línea de reproducción,
vi) nombre vulgar;
b) distribución geográfica y cultivo de la planta en la Unión;
c) información sobre la reproducción:
i) modo o modos de reproducción,
ii) de haberlos, factores específicos que afecten a la reproducción,
iii) período de generación;
d) compatibilidad sexual con otras especies vegetales cultivadas o silvestres, indicando la distribución de las especies compatibles en la Unión;
e) capacidad de supervivencia:
i) capacidad para formar estructuras de supervivencia o de latencia,
ii) de haberlos, factores específicos que afecten a la supervivencia;
f) diseminación:
i) formas y alcance de la diseminación,
ii) de haberlos, factores específicos que afecten a la diseminación;
g) en el caso de especies vegetales que no crezcan normalmente en la Unión, una descripción del hábitat natural de la planta que incluya información sobre predadores naturales, parásitos, competidores y simbiontes;
h) posibles interacciones de la planta, pertinentes para la PSMG, con organismos presentes en el ecosistema en el que esta crece normalmente, o en otros lugares, incluida información sobre los efectos tóxicos para las personas, los animales y otros organismos.
2. Caracterización molecular.
a) Información relativa a la modificación genética:
i) descripción de los métodos utilizados para la modificación genética,
ii) naturaleza y origen del vector utilizado,
iii) origen del ácido nucleico o los ácidos nucleicos utilizado(s) para la transformación, tamaño y función prevista de cada fragmento componente de la región que se pretenda insertar.
b) Información relativa a la planta modificada genéticamente:
i) descripción de los rasgos y características que se han introducido o modificado,
ii) información sobre las secuencias realmente insertadas o suprimidas:
– el tamaño y el número de copias de todos los insertos detectables, tanto completos como parciales, y los métodos empleados para su caracterización,
– la organización y secuencia del material genético insertado en cada sede de inserción en un formato electrónico normalizado,
– en caso de supresión, tamaño y función de la región o regiones suprimidas,
– localización subcelular del inserto o los insertos (integrados en el núcleo, cloroplastos o mitocondrias, o mantenidos en una forma no integrada), y métodos para su determinación,
– en caso de modificaciones distintas de una inserción o una supresión, la función del material genético modificado antes y después de la modificación, así como los cambios directos en la expresión de genes como resultado de la modificación,
– secuencia de información en un formato electrónico normalizado para las regiones flanqueadoras de 5′ y 3′ en cada sede de inserción,
– análisis bioinformático utilizando bases de datos actualizadas, con objeto de investigar posibles interrupciones de genes conocidos,
– todos los marcos abiertos de lectura (en lo sucesivo, denominados «MAL») en el inserto (sean o no resultado de un reajuste), así como los creados como resultado de la modificación genética en los sitios de unión con ADN genómico; un MAL se define como una secuencia de nucleótidos que contiene una hilera de codones no interrumpida por la presencia de un codón de terminación en el mismo marco de lectura,
– análisis bioinformático utilizando bases de datos actualizadas, con objeto de investigar posibles similitudes entre los MAL y genes conocidos que pueden tener efectos adversos,
– estructura primaria (secuencia de aminoácidos) y, en su caso, otras estructuras, de la proteína de nueva expresión,
– análisis bioinformático utilizando bases de datos actualizadas, con objeto de investigar posibles homologías de secuencia y, en su caso, semejanzas estructurales entre las proteínas de nueva expresión y las proteínas conocidas o los péptidos que pueden tener efectos adversos,
iii) información sobre la expresión del inserto:
– método o métodos utilizados para el análisis de expresión junto con sus características de rendimiento,
– información sobre la expresión del desarrollo del inserto durante el ciclo biológico de la planta,
– partes de la planta en que se expresan el fragmento de inserción/la secuencia modificada,
– posible expresión no intencional de nuevos MAL identificados con arreglo al inciso séptimo del inciso ii), que planteen un problema de seguridad,
– datos sobre la expresión de las proteínas, incluidos los datos brutos, obtenidos a partir de estudios de campo y relacionados con las condiciones de cultivo del vegetal,
iv) estabilidad genética del inserto y estabilidad fenotípica de la PSMG:
c) Conclusiones de la caracterización molecular:
3. Análisis comparativo de las características agronómicas y fenotípicas y de la composición.
a) elección del homólogo convencional y otros referentes de comparación;
b) selección de emplazamientos para los estudios de campo;
c) diseño experimental y análisis estadístico de los datos procedentes de ensayos de campo para el análisis comparativo:
i) descripción del diseño de los estudios de campo,
ii) descripción del aspecto pertinente de los entornos receptores,
iii) análisis estadístico;
d) si es pertinente, selección de material vegetal para el análisis;
e) análisis comparativo de las características agronómicas y fenotípicas;
f) análisis comparativo de la composición, si resulta pertinente;
g) conclusiones del análisis comparativo.
4. Información específica para cada ámbito de riesgo.
Para cada uno de los siete ámbitos de riesgo mencionados en la sección D.2 del anexo IV, el notificador describirá primero la vía perjudicial, explicando, en una cadena de causa-efecto, cómo la liberación de la PSMG puede resultar perjudicial, habida cuenta del peligro y de la exposición.
El notificador presentará la siguiente información, excepto si esta no es pertinente a la luz de los usos previstos del OMG:
a) Persistencia y capacidad de invasión, incluida la transferencia de material genético entre plantas:
i) evaluación de las posibilidades de que la PSMG se vuelva más persistente o invasiva y los efectos medioambientales adversos que se derivan,
ii) evaluación de las posibilidades de que la PSMG transmita transgenes a variedades emparentadas sexualmente compatibles y los efectos medioambientales adversos que se derivan,
iii) conclusiones sobre los efectos ambientales adversos de la persistencia y la capacidad de invasión de la PSMG, incluidos los efectos adversos para el medio ambiente de la transferencia de material genético entre plantas;
b) Transferencia genética de las plantas a los microorganismos:
i) evaluación de las posibilidades de transferencia del ADN recién insertado de la PSMG a microorganismos y los efectos medioambientales adversos que se derivan,
ii) conclusiones sobre los efectos adversos de la transferencia de ADN recién insertado de la PSMG a los microorganismos para la salud humana y animal y para el medio ambiente;
c) Interacciones de la PSMG con los organismos objetivo, en su caso:
i) evaluación del potencial de cambio en las interacciones directas e indirectas entre la PSMG y los organismos objetivo y los efectos adversos en el medio ambiente,
ii) evaluación del potencial de evolución de la resistencia del organismo objetivo a la proteína expresada (basándose en los antecedentes de la evolución de la resistencia a los plaguicidas convencionales o las plantas transgénicas con rasgos similares) y los efectos adversos para el medio ambiente que se derivan,
iii) conclusiones sobre los efectos adversos para el medio ambiente de las interacciones entre la PSMG y los organismos objetivo;
d) Interacciones de la PSMG con organismos no objetivo:
i) evaluación del potencial de que se produzcan interacciones directas e indirectas entre la PSMG y organismos no objetivo, incluidas las especies protegidas, y los efectos adversos que se derivan.
La evaluación tendrá asimismo en cuenta los posibles efectos adversos sobre los servicios ecosistémicos pertinentes y sobre las especies responsables de esos servicios,
ii) conclusiones sobre los efectos adversos para el medio ambiente de las interacciones entre la PSMG y organismos no objetivo;
e) Impacto de las técnicas específicas de cultivo, gestión y recolección:
i) en relación con las PSMG para cultivo, evaluación de los cambios en las técnicas específicas de cultivo, gestión y recolección utilizadas para la PSMG en cuestión y los efectos adversos para el medio ambiente que se derivan,
ii) conclusiones sobre los efectos adversos para el medio ambiente de las técnicas específicas de cultivo, gestión y recolección;
f) Efectos sobre los procesos biogeoquímicos:
i) evaluación de los cambios en los procesos biogeoquímicos en la zona en la que se cultivará la PSMG y en el medio ambiente en general, así como los efectos adversos que se derivan,
ii) conclusiones sobre los efectos adversos en los procesos biogeoquímicos;
g) Efectos en la salud humana y animal:
i) evaluación de las posibles interacciones directas o indirectas entre la PSGM y las personas que manejan PSMG o están en contacto con estas, incluso a través del polen o el polvo de una PSMG transformada, y evaluación de los efectos adversos de estas interacciones en la salud humana,
ii) por lo que se refiere a las PSMG no destinadas al consumo humano, pero cuyos organismos receptores o parentales pueden destinarse al consumo humano, evaluación de la probabilidad de que se produzca una ingesta accidental y de sus posibles efectos adversos en la salud humana,
iii) evaluación de los posibles efectos adversos sobre la salud de los animales, derivados de un consumo accidental, por parte de los animales, de la PSMG o de material procedente de dicha planta,
iv) efectos en la salud humana y de los animales;
h) Evaluación global del riesgo y conclusiones:
Se suministrará un resumen de todas las conclusiones en cada ámbito de riesgo.
El resumen deberá tener en cuenta la caracterización del riesgo de conformidad con las fases 1 a 4 de la metodología descrita en la sección C.3 del anexo IV y las estrategias de gestión del riesgo propuestas de conformidad con el punto 5 de la sección C.3 del anexo IV.
5. Descripción de las técnicas de detección e identificación de la PSMG.
6. Información sobre anteriores liberaciones de la PSMG, en su caso.
Se modifica por el art. 2.18 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Esta modificación entra en vigor el 30 de septiembre de 2019, según establece la disposición final 2 del citado Real Decreto.
[Bloque 100: #anvi]
(Artículo 28)
Se enumeran a continuación los criterios a que se refiere el artículo 28.
1. La taxonomía y la biología (modo de reproducción y polinización, capacidad de cruzarse con especies afines, patogenia) del organismo (receptor) no modificado deberán conocerse bien.
2. La seguridad para la salud humana y el medio ambiente de los organismos parental, cuando proceda, y receptor en el entorno de la liberación deberá conocerse en suficiente medida.
3. Deberá haber información disponible sobre cualquier interacción de particular relevancia para la evaluación del riesgo que implique a los organismos parental, cuando proceda, y receptor y a otros organismos en el ecosistema de liberación experimental.
4. Deberá haber información disponible que demuestre que todo material genético introducido está bien caracterizado, así como información sobre la construcción de sistemas de vectores o de secuencias de material genético empleados con el ADN portador. Cuando una modificación genética incluya la supresión de material genético, deberá conocerse la amplitud de la supresión. Asimismo deberá aportarse información suficiente sobre la modificación genética para permitir la identificación del organismo modificado genéticamente y su progenie durante una liberación.
5. El organismo modificado genéticamente no deberá presentar riesgos mayores ni más numerosos para la salud humana o para el medio ambiente en las condiciones de liberación experimental que los presentes en las liberaciones de los organismos parentales, cuando proceda, y receptores correspondientes. Ninguna capacidad de propagación en el medio ambiente, de invasión de ecosistemas no relacionados y de transferir material genético a otros organismos en el medio ambiente deberá acarrear efectos adversos.
[Bloque 101: #anvii]
1. Las solicitudes de autorización y, en especial, la información a suministrar se regirán por lo establecido en el capítulo II.
2. Para obtener una autorización única que incluya a varias liberaciones, se deberá facilitar en la solicitud toda la información necesaria sobre cada una de ellas, incluyendo los distintos lugares de liberación y el diseño experimental, así como la indicación de cualquier condición de gestión de riesgo para cada liberación en particular. Se deberá hacer una referencia clara a cada liberación incluida en la solicitud y adjuntar la información adecuada para poder completar el modelo resumido de solicitud e información.
3. En los supuestos regulados en el artículo 29, no será necesario proporcionar en la solicitud indicaciones ni descripciones detalladas de los lugares de liberación, de los subsiguientes cruces sexuales intraespecíficos, ni de las condiciones de liberación. No obstante, en la solicitud se deberán exponer suficientes datos para que se pueda realizar una evaluación global de los riesgos y una evaluación pormenorizada, que se hará al menos para la primera liberación incluida en el programa de trabajo. Cuando sea de aplicación lo establecido en el apartado anterior, el solicitante presentará, en su caso, al órgano competente información adicional junto con una declaración en la que se indique si continúa siendo válida la evaluación de riesgo original y, en caso de que no lo sea, facilitará una nueva evaluación. Esta información se enviará antes de proceder a la liberación específica a la que se refiere, en forma de una simple notificación adicional a efectos solamente informativos.
En estos casos, el órgano competente enviará inmediatamente a la Comisión Europea cualquier información adicional sobre la evaluación del riesgo recibida en aplicación de lo establecido en el párrafo anterior.
Si la información adicional presentada mostrara que la autorización original por procedimiento simplificado ha dejado de ser aplicable, el órgano competente deberá indicar al solicitante, en el plazo de 15 días a partir de la recepción de la notificación, que sólo podrá proceder a la liberación propuesta si obtiene una autorización por el procedimiento normal establecido en este reglamento. Transcurrido este plazo sin que el órgano competente haya adoptado ninguna resolución al respecto, el solicitante podrá continuar con la liberación específica de que se trate.
4. Cuando se conceda la autorización única en los términos del procedimiento simplificado, se pueden poner condiciones a cada una de las liberaciones a las que se refiera. Estas condiciones podrán ser modificadas por el órgano competente, de acuerdo con lo establecido en los artículos 46.2 y 47.1.
5. Una vez realizadas una o varias de las liberaciones aprobadas en virtud del procedimiento simplificado, el solicitante presentará al órgano competente un informe con los resultados de una o varias liberaciones en el plazo que se indique en la autorización. Dichos informes podrán presentarse por separado o como parte claramente identificable de una notificación de liberaciones subsiguientes.
6. El órgano competente podrá alterar las condiciones de la primera autorización o intervenir para alterar las condiciones de liberaciones específicas subsiguientes, basándose en los resultados de los informes o en la información obtenida en el curso de inspecciones.
[Bloque 102: #anviii]
En este anexo se describe en términos generales la información complementaria que deberá aportarse en caso de solicitud de autorización de comercialización, así como la información exigida para el etiquetado del organismo modificado genéticamente como producto o componente del producto que se comercializa y del organismo modificado genéticamente utilizado en operaciones que no se consideran comercialización en virtud del artículo 30.2. Se complementará con notas orientativas en lo relativo, por ejemplo, a la descripción del uso previsto para el producto, que se aprueben en la normativa comunitaria. El etiquetado de los organismos exceptuados contemplado en el artículo 50.3 se establecerá mediante la formulación de las adecuadas recomendaciones y restricciones de su uso. Como complemento a este anexo deberán aplicarse los requisitos que establezca la legislación comunitaria en relación con la información sobre las modificaciones genéticas que deberá figurar en el registro que se menciona el apartado A 7.
A.
En la solicitud de autorización de comercialización de un organismo modificado genéticamente como producto o componente de un producto, deberá aportarse, además de la exigida en el anexo V, la siguiente información:
1. Las propuestas de nombres comerciales para los productos y los nombres de los OMG que contengan, así como una propuesta de identificador único del OMG, elaboradas de conformidad con el Reglamento (CE) n.º 65/2004 de la Comisión, de 14 de enero de 2004, por el que se establece un sistema de creación y asignación de identificadores únicos a los organismos modificados genéticamente. Una vez concedida la autorización, todos los nuevos nombres comerciales deberán aportarse a la autoridad competente.
2. El nombre y la dirección completa de la persona domiciliada en la Comunidad Europea responsable de la comercialización, sea el fabricante, el importador o el distribuidor.
3. El nombre y la dirección completa del suministrador o de los suministradores de las muestras de control.
4. La descripción de cómo se prevé que se utilice el producto y el organismo modificado genéticamente como producto o componente del producto. Deberán resaltarse las diferencias de uso o gestión del organismo modificado genéticamente en comparación con productos similares no modificados genéticamente.
5. La descripción de la zona o de las zonas geográficas y de los tipos de entorno en que se prevé el uso del producto dentro de la Comunidad Europea, incluida, cuando sea posible, una estimación de la escala de su uso en cada zona.
6. Las categorías de usuarios previstas para el producto, como por ejemplo industria, agricultura o artesanía o consumo por parte del público en general.
7. Métodos de detección, identificación y, en su caso, cuantificación del evento de transformación; muestras del o de los OMG y sus muestras de control, e información sobre el lugar en que se puede acceder al material de referencia. Deberá identificarse la información que no deba figurar, por motivos de confidencialidad, en la parte accesible al público del registro.
8. La propuesta de etiquetado, sobre una etiqueta o en un documento adjunto, que debe incluir, al menos en forma de síntesis, un nombre comercial para el producto, la mención «este producto contiene organismos modificados genéticamente», el nombre del organismo modificado genéticamente y la información a que se refiere el apartado A 2. El etiquetado deberá indicar la manera de acceder a la información recogida en la parte del registro accesible al público.
B.
Como complemento de lo estipulado en el apartado A, deberá aportarse, conforme a lo dispuesto en el artículo 30, la siguiente información:
1. Las medidas que deberán tomarse en caso de liberación involuntaria o utilización incorrecta del producto.
2. Las instrucciones específicas o recomendaciones para su almacenamiento y manipulación.
3. Las instrucciones específicas para el control e información al interesado y, en su caso, al órgano competente, de tal manera que se informe eficazmente a las autoridades competentes de cualquier efecto adverso.
Estas instrucciones deberán ser compatibles con el apartado C del anexo X.
4. Las propuestas de restricciones para la utilización aprobada del organismo modificado genéticamente, por ejemplo, dónde puede utilizarse el producto y con qué fines.
5. El envasado propuesto.
6. Una estimación de la producción interior y/o de la importación a la Comunidad Europea.
7. Una propuesta de etiquetado complementario, que podrá incluir, al menos en forma de síntesis, la información a que se refieren el apartado A 4 y 5 y el apartado B 1, 2, 3 y 4.
Se modifica la Sección A por el art. 2.19 del Real Decreto 452/2019, de 19 de julio. Ref. BOE-A-2019-10757
Esta modificación entra en vigor el 30 de septiembre de 2019, según establece la disposición final 2 del citado Real Decreto
[Bloque 103: #anix]
El informe de evaluación previsto en los artículos 34. 3, 41.1 y 44 2, deberá incluir, en particular, lo siguiente:
1. La identificación de las características del organismo receptor pertinentes para la evaluación del organismo o de los organismos modificados genéticamente en cuestión. La identificación de cualquier riesgo conocido para la salud humana y el medio ambiente resultante de la liberación del organismo receptor no modificado en el medio ambiente.
2. La descripción del resultado de la modificación genética en el organismo modificado.
3. Una evaluación de si la identificación de las características de la modificación genética es suficiente con el fin de evaluar los riesgos para la salud humana y el medio ambiente.
4. La identificación de todo nuevo riesgo para la salud humana y para el medio ambiente que se pueda producir a partir de la liberación del organismo o de los organismos modificados genéticamente en cuestión comparado con la liberación del organismo o de los organismos no modificados correspondientes basado en la evaluación del riesgo para la salud humana el medio ambiente efectuada de acuerdo con lo dispuesto en el anexo IV.
5. Una conclusión sobre si el o los organismos modificados genéticamente en cuestión deberán comercializarse como producto(s) o componente(s) de un producto(s) y en qué condiciones, si el organismo o los organismos modificados genéticamente en cuestión no pueden comercializarse o si se requiere el parecer de otras autoridades competentes y de la Comisión Europea en cuestiones específicas de la evaluación del riesgo para la salud humana y el medio ambiente. Estos aspectos deberán especificarse. La conclusión deberá referirse claramente al uso propuesto, a la gestión del riesgo y al plan de seguimiento propuesto. En caso de que se llegue a la conclusión de que el organismo o los organismos modificados genéticamente en cuestión no deben comercializarse, el Consejo interministerial de organismos modificados genéticamente motivará su conclusión.
[Bloque 104: #anx]
Este anexo describe de modo general el objetivo que debe lograrse y los principios generales que deben seguirse para diseñar el plan de seguimiento mencionado en el artículo 32.2, en el artículo 37 y en el artículo 42. Como complemento a este anexo deberán utilizarse las notas de orientación establecidas en el anexo de la Decisión 2002/811/CE del Consejo, de 3 de octubre de 2002, y cumplir cualesquiera otras disposiciones comunitarias que se aprueben en la materia, bien sean de aplicación directa, bien una vez que se produzca su incorporación al ordenamiento interno.
A. Objetivo
El objetivo de un plan de seguimiento es:
1.° Confirmar que cualquier suposición relativa a que se produzcan y a las consecuencias de efectos adversos potenciales del organismo modificado genéticamente o de su uso en la evaluación del riesgo para la salud humana y el medio ambiente son correctos, e
2.° Identificar que se produzcan efectos adversos del organismo modificado genéticamente o de su uso en la salud humana o el medio ambiente que no se hayan contemplado en la evaluación del riesgo para la salud humana y el medio ambiente.
B. Principios generales
El seguimiento, conforme a lo dispuesto en los artículos 32.2, 37 y 42, está teniendo lugar a raíz de la autorización para comercializar un organismo modificado genéticamente.
La interpretación de los datos recogidos por el seguimiento debería considerarse a la luz de otras condiciones medioambientales y actividades existentes. Cuando se observen cambios en el medio ambiente, se deberá considerar una nueva evaluación que establezca si son una consecuencia del organismo modificado genéticamente o de su uso, ya que tales cambios pueden ser el resultado de factores medioambientales distintos de la comercialización del organismo modificado genéticamente.
La experiencia y los datos adquiridos a través del seguimiento de liberaciones experimentales de organismos modificados genéticamente pueden ayudar a diseñar el régimen de seguimiento posterior a la investigación del mercado requerida para la comercialización de los organismos modificados genéticamente como productos o como componentes de productos.
C. Diseño del plan de seguimiento
El diseño del plan de seguimiento deberá:
1. Detallarse en cada caso teniendo en cuenta la evaluación del riesgo para la salud humana y el medio ambiente.
2. Tener en cuenta las características del organismo modificado genéticamente, las características y la escala de su uso previsto y la gama de condiciones medioambientales pertinentes donde se espera que se libere el organismo modificado genéticamente.
3. Incluir una vigilancia general para detectar los efectos adversos imprevistos, así como, en caso necesario, un control especifico (para determinados casos) centrado en los efectos adversos identificados en la evaluación del riesgo para la salud humana y el medio ambiente:
3.1 Considerando que el seguimiento específico de cada caso deberá llevarse a cabo durante un plazo suficiente para detectar los efectos inmediatos y directos, así como, si procede, los efectos diferidos o indirectos que se hayan identificado en la evaluación del riesgo para la salud humana y el medio ambiente.
3.2 Considerando que la vigilancia podrá hacer uso, si procede, de prácticas rutinarias de vigilancia ya establecidas, tales como la supervisión de cultivos agrícolas, la protección fitosanitaria, o de productos veterinarios y médicos. Se explicará al titular de la autorización la manera en que la información pertinente recogida mediante prácticas rutinarias de vigilancia ya establecidas se pondrá a su disposición.
4. Facilitar la observación, de manera sistemática, de la liberación de un organismo modificado genéticamente en el entorno receptor y la interpretación de dichas observaciones en lo que se refiere a la seguridad de la salud humana o del medio ambiente.
5. Determinar quién llevará a cabo las diversas tareas que requiere el plan de seguimiento y quién es responsable de asegurar que el plan de seguimiento se plantee y se lleve a cabo debidamente, y de asegurar que haya un canal que permita que el titular de la autorización y el Consejo interministerial de organismos modificados genéticamente estén informados sobre cualquier efecto adverso que se observe en la salud humana y el medio ambiente. (Se indicarán las fechas y los intervalos de tiempo para los informes sobre los resultados de la supervisión).
6. Considerar los mecanismos para identificar y confirmar cualquier efecto adverso que se observe en la salud humana y el medio ambiente y permitir que el titular de la autorización o la autoridad competente, en su caso, tomen las medidas necesarias para proteger la salud humana y el medio ambiente.
[Bloque 105: #anxi]
El titular de la actividad utilizará el "modelo de informe" que figura a continuación para la presentación al organismo competente de los resultados de la liberación intencional en el medio ambiente de plantas superiores modificadas genéticamente.
Cada formulario corresponderá a una autorización concedida y se identificará por su número de notificación.
Para cada número de notificación el titular presentará un informe final y, en su caso, uno o varios informes intermedio de seguimiento posterior a la liberación. Ambos informes se ajustarán al modelo de informe.
El informe final deberá entregarse después de la última cosecha. Este será el único informe en caso de que la liberación no requiera un seguimiento posterior.
El organismo competente precisará en su autorización, cuando proceda, la duración del seguimiento posterior a la liberación, así como la periodicidad de la presentación de los informes intermedios del seguimiento posterior a la liberación.

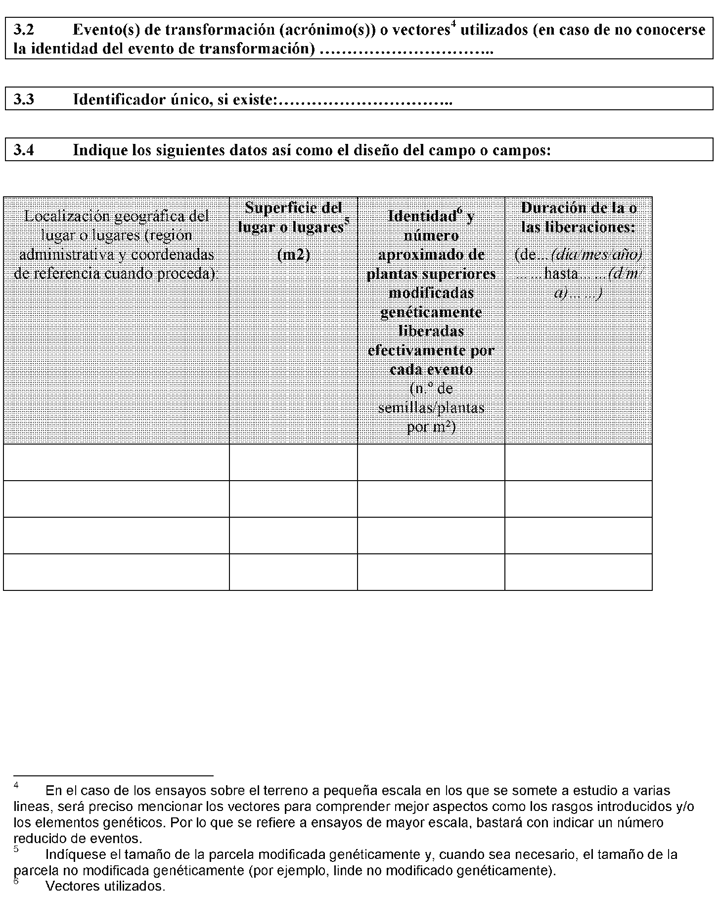








[Bloque 106: #informacionrelacionada]
Información relacionada
Téngase en cuenta las referencias hechas a determinados órganos administrativos, según establece la disposición adicional única del Real Decreto 406/2021, de 8 de junio. Ref. BOE-A-2021-10229
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid





