Agencia Estatal Boletín Oficial del Estado






 Este texto consolidado es de carácter informativo y no tiene valor jurídico.
Este texto consolidado es de carácter informativo y no tiene valor jurídico.[Disposición derogada]
Norma derogada por la disposición derogatoria única del Real Decreto 600/2016, de 2 de diciembre. Ref. BOE-A-2016-11480.
[Bloque 2: #preambulo]
Siendo necesario adecuar la legislación española a lo dispuesto en la Directiva del Consejo 83/417 de la Comunidad Económica Europea relativa a la aproximación de las legislaciones de los Estados miembros sobre determinadas lactoproteínas (caseínas y caseinatos) destinadas a la alimentación humana, así como a las Directivas de la Comisión 85/503 y 86/424, relativas a los métodos de análisis y a los procedimientos comunitarios de toma de muestras destinadas a análisis químicos para caseínas y caseinatos, parece oportuno dictar las presentes Normas Generales de Calidad para Caseínas y Caseinatos Alimenticios.
En su virtud, oídos los sectores afectados, previo informe preceptivo de la Comisión Interministerial para la Ordenación Alimentaria, y a propuesta de los Ministros de Economía y Hacienda, de Agricultura, Pesca y Alimentación, y de Sanidad y Consumo, este Ministerio de Relaciones con las Cortes y de la Secretaría del Gobierno dispone:
[Bloque 3: #primero]
Se aprueban las Normas Generales de Calidad para Caseínas y Caseinatos Alimenticios con destino al mercado interior, que se recogen, respectivamente, en los anejos 1 y 2 de esta Orden.
[Bloque 4: #segundo]
La toma de muestras y las determinaciones analíticas se realizarán de acuerdo con los métodos que se recogen en los anejos 3 y 4 de esta Orden.
[Bloque 5: #tercero]
Los Departamentos competentes velarán por el cumplimiento de lo dispuesto en la presente Orden a través de sus órganos administrativos encargados, que coordinarán sus actuaciones, en todo caso, sin perjuicio de las competencias que correspondan a las Comunidades Autónomas y a las Corporaciones Locales.
[Bloque 6: #cuao]
La presente Orden entrará en vigor el día siguiente al de su publicación en el «Boletín Oficial del Estado».
[Bloque 7: #firma]
Madrid, 28 de marzo de 1988.
ZAPATERO GÓMEZ
Excmos. Sres. Ministros de Economía y Hacienda, Agricultura, Pesca y Alimentación y de Sanidad y Consumo.
[Bloque 8: #anejo1]
1. Nombre de la norma.
Norma general de calidad para las caseínas alimenticias.
2. Objeto de la norma.
La presente norma tiene por objeto definir aquellas condiciones y características que deben reunir las caseínas alimenticias para su comercialización y utilización en el mercado interior.
3. Ámbito de aplicación.
La presente norma general abarca a todas las caseínas alimenticias destinadas a su comercialización en el mercado interior para uso en la industria alimenticia.
Los productos definidos en esta norma sólo pueden comercializarse si se ajustan a las definiciones y requisitos previstos en ella.
4. Definiciones.
4.1 Caseína: A los efectos de la presente norma se entiende por «caseínas» el principal componente proteico de la leche, insoluble en el agua obtenido por lavado, prensado y secado del coágulo obtenido a partir de leche desnatada por adición de ácido, o por acidificación microbiana (caseína ácida), o por medio de cuajo o de otros enzimas coagulantes de leche (caseína al cuajo), sin perjuicio de una posible aplicación previa de procedimientos de intercambios de iones y de concentración.
4.2 Caseínas alimenticias: Son aquellas caseínas para cuya elaboración se somete obligatoriamente a un proceso de pasterización la leche desnatada utilizada, el coágulo obtenido de la misma, o la suspensión acuosa de la caseína.
5. Denominaciones.
Las denominaciones que figuran a continuación quedan reservadas a los productos objeto de la norma y deberán utilizarse obligatoriamente en la industrialización y comercialización de los mismos.
5.1 Caseína ácida alimenticia: Se denomina así a la caseína alimenticia para cuya obtención se han utilizado los coadyuvantes tecnológicos y/o cultivos bacterianos enumerados en el apartado 6.3.1, que se ajuste a los requisitos establecidos en el apartado 6.
5.2 Caseína al cuajo alimenticia: Se denomina así a la caseína alimenticia para cuya obtención se han utilizado los coadyuvantes tecnológicos enumerados en el apartado 6.3.2, que se ajuste a los requisitos establecidos en el apartado 6.
6. Factores esenciales de composición y calidad.
6.1 Ingredientes esenciales: Leche desnatada.
6.2 Características físico-químicas:
|
|
Caseína ácida alimenticia – Porcentaje |
Caseína al cuajo alimenticia – Porcentaje |
|---|---|---|
|
Contenido máximo de humedad |
10 m/m |
10 m/m |
|
Contenido mínimo de proteínas de la leche calculado sobre extracto seco |
90 m/m |
84 m/m |
|
Contenido mínimo de caseínas sobre proteinas lácteas |
95 m/m |
95 m/m |
|
Contenido máximo de matería grasa de la leche sobre extracto seco |
2,25 m/m |
2 m/m |
|
Acidez titulable máxima expresada en ml de solución de hidróxido sódico decinormal por gramo |
0,27 ml/g |
|
|
Contenido de cenizas (P2O5 incluido) |
(máx.) 2,5 m/m |
(min.) 7,5 m/m |
|
Contenido máximo de lactosa anhidra |
1 m/m |
1 m/m |
|
Contenido máximo de sedimentos (partículas quemadas) |
22,5 mg en 25 g |
22,5 mg en 25 g |
|
Prueba de la fosfatasa |
Negativa |
Negativa |
6.3 Coadyuvantes tecnológicos:
6.3.1 Para la caseína ácida alimenticia:
Ácido láctico (E 270).
Ácido clorhídrico.
Ácido sulfúrico.
Ácido cítrico (E 330).
Ácido acético (E 260).
Ácido ortofosfórico (E 338).
Lactosuero.
Cultivos bacterianos que produzcan ácido láctico (fermentos lácticos).
6.3.2 Para la caseína al cuajo alimenticia:
Cuajo.
Otros enzimas coagulantes de la leche.
6.4 Caracteres organolépticos: Olor: Ausencia de olores extraños.
Aspecto: Color del blanco al blanco crema; el producto deberá estar exento de grumos que resistan una presión ligera.
7. Contaminantes.
(Derogado)
8. Prohibiciones.
Queda expresamente prohibido:
8.1 (Derogado)
8.2 La presencia en las caseínas alimenticias de proteínas y/o grasas distintas a las de la propia leche.
8.3 La comercialización y venta de productos que no ajustándose a la presente norma incluyan en su denominación las palabras «caseína ácida alimenticia» o «caseína al cuajo alimenticia».
8.4 (Derogado)
9. Higiene.
9.1 La leche desnatada, el coágulo de ella obtenido o la suspensión acuosa de caseína deben de someterse a un tratamiento térmico equivalente, al menos, a la pasterización. Es decir, que la prueba de la fosfatasa debe de dar negativa.
9.2 a 3 (Derogados)
10. Envasado.
(Derogado)
11. Etiquetado.
Los datos obligatorios que figuren en el etiquetado de los envases deberán ser fácilmente comprensibles e irán inscritos en un lugar destacado y de forma que sean fácilmente visibles, claramente legibles e indelebles. Esta información no deberá ser enmascarada por dibujos ni por cualquier texto o imagen, escrito, impreso o gráfico.
Los datos obligatorios del etiquetado de los productos alimenticios que se comercialicen en España se expresarán necesariamente al menos en la lengua española oficial del Estado.
La información del etiquetado de los envases de las caseínas alimenticias sujetas a esta norma de calidad constará obligatoriamente de las siguientes especificaciones:
11.1 Denominación del producto:
a) Se indicará conforme al apartado 5 de esta norma.
b) Cuando se comercialicen caseínas alimenticias en mezcla simple con otros productos, la denominación será:
La mención «Mezcla de ....................... » seguida de las denominaciones de los diferentes productos que constituyan la mezcla, en orden ponderal decreciente.
La indicación del o de los cationes para el o los caseinatos.
La proporción de proteínas para las mezclas que contengan caseinatos.
11.2 a 4 (Derogados)
12. Rotulación.
(Derogado)
13. Responsabilidades.
(Derogado)
14. Régimen sancionador.
(Derogado)
Se derogan los apartados 7, 8.1, 8.4, 9.2, 9.3, 10, 11 excepto subapartado1,12,13 y 14 por el art. 44.1 del Real Decreto 176/2013, de 8 de marzo. Ref. BOE-A-2013-3402.
[Bloque 9: #anejo2]
1. Nombre de la norma
Norma general de calidad para los caseinatos alimenticios.
2. Objeto de la norma
La presente norma tiene por objeto definir aquellas condiciones y características que deben reunir los caseinatos alimenticios para su comercialización y utilización en el mercado interior.
3. Ámbito de aplicación
La presente norma general abarca a todos los caseinatos alimenticios destinados a su comercialización en el mercado interior para uso en la industria alimenticia.
Los productos definidos en esta norma sólo pueden comercializarse si se ajustan a las definiciones y requisitos previstos en ella.
4. Definiciones
4.1 Caseinatos: Se entiende por caseinatos los productos obtenidos por el secado de caseínas en suspensión acuosa tratadas con uno o más agentes neutralizados.
4.2 Caseinatos alimenticios: Se entiende por caseinatos alimenticios los caseinatos obtenidos a partir de caseínas alimenticias en suspensión acuosa, tratadas con uno o más de los coadyuvantes tecnológicos que figuran en el apartado 6.3 y que respondan a los requisitos establecidos en el apartado 6.
5. Denominaciones
Los caseinatos alimenticios se denominarán «Caseinato de ... alimenticio», incluyendo el nombre del catión o cationes, en este último caso separados por una «y», o bien «Caseinato ... alimenticio», incluyendo el nombre adjetivado del catión o cationes.
Dichas denominaciones quedan reservadas a los productos objeto de esta norma y deberán utilizarse obligatoriamente en la industrialización y comercialización de los mismos.
6. Factores esenciales de composición y calidad
6.1 Ingredientes esenciales: Caseínas alimenticias.
6.2 Características físico-químicas:
Contenido máximo de humedad: 8 % m/m.
Contenido mínimo de caseína proteica de la leche, calculada sobre extracto seco: 88% m/m.
Contenido máximo de materias grasas de la leche calculado sobre extracto seco: 2% m/m.
Contenido máximo de lactosa anhidra: 1 % m/m.
pH: 6 a 8.
Contenido máximo de sedimentos (partículas quemadas): 22,5 mg en 25 g.
Prueba de la fosfatasa: Negativa.
Los caseinatos son casi totalmente solubles en agua destilada, a excepción del caseinato de calcio que puede presentar distintos grados de solubilidad.
6.3 Coadyuvantes tecnológicos de calidad alimenticia: Pueden utilizarse los siguientes agentes neutralizantes y tampones opcionales:
Hidróxidos de sodio.
Carbonatos de potasio.
Fosfatos de calcio.
Fosfatos de amonio.
Citratos de magnesio.
6.4 Caracteres organolépticos:
Olor: Aroma y olores extraños ligeros.
Aspecto: Color del blanco al blanco crema; el producto deberá estar exento de grumos que resistan una presión ligera.
7. Contaminantes
(Derogado)
8. Prohibiciones
Queda expresamente prohibido:
8.1 (Derogado)
8.2 La presencia en los caseinatos alimenticios de proteínas y/o grasas distintas a las de la propia leche.
8.3 La comercialización y venta de productos que no ajustándose a la presente norma incluyan en su denominación las palabras «caseinato alimenticio».
8.4 (Derogado)
9. Higiene
(Derogado)
10. Envasado
(Derogado)
11. Etiquetado
Los datos obligatorios que figuren en el etiquetado de los envases deberán ser fácilmente comprensibles e irán inscritos en un lugar destacado y de forma que sean fácilmente visibles, claramente legibles e indelebles. Esta información no deberá ser enmascarada por dibujos, ni por cualquier texto o imagen, escrito, impreso o gráfico.
Los datos obligatorios del etiquetado de los productos alimenticios que se comercialicen en España se expresarán necesariamente al menos en la lengua española oficial del Estado.
La información del etiquetado de los envasés de los caseinatos alimenticios sujetos a esta norma de calidad constará obligatoriamente de las siguientes especificaciones:
11.1 Denominación del producto:
a) Se indicará conforme al apartado 5 de esta norma.
b) Cuando se comercialicen caseinatos alimenticios en mezcla simple con otros productos, la denominación será:
La mención «Mezcla de ........» seguida de las denominaciones de los diferentes productos que constituyan la mezcla, en orden ponderal decreciente.
La indicación del o de los cationes para el o los caseinatos.
La proporción de proteínas.
11.2 a 6 (Derogados)
12. Rotulación
(Derogado)
13. Responsabilidades
(Derogado)
14. Régimen sancionador
(Derogado)
Se derogan los apartados 7, 8.1, 8.4, 9, 10, 11 excepto subapartado1,12,13 y 14 por el art. 44.2 del Real Decreto 176/2013, de 8 de marzo. Ref. BOE-A-2013-3402.
[Bloque 10: #anejo3]
I. DISPOSICIONES GENERALES
1. Prescripciones administrativas
1.1 Personal: La toma de muestras la llevará a cabo una persona autorizada y cualificada, tal como establezcan las disposiciones en vigor en los Estados miembros.
1.2 Sellado y etiquetado de las muestras: Cada muestra oficial se cerrará, se sellará en el lugar de la toma y se identificará según las disposiciones en vigor en los Estados miembros.
1.3 Muestras duplicadas: Se prepararán sumultáneamente para el análisis al menos dos muestras equivalentes y representativas. Sin perjuicio de las disposiciones comunitarias que pudieran adoptarse, el número de muestras que deberá tomarse estará en función de la legislación nacional aplicable en la materia en cada Estado miembro. Una vez efectuada la toma, se remitirán las muestras al laboratorio tan pronto como sea posible.
1.4 Informe: A las muestras se acompañará un informe, el cual se elaborará según la legislación de los Estados miembros.
2. Equipo de toma de muestras
Características:
El equipo de toma de muestras no deberá provocar ningún cambio en la muestra que pudiera alterar los resultados de los análisis, y deberá ser suficientemente resistente, con objeto de evitar cualquier falseamiento de los resultados. Se recomienda utilizar material de acero inoxidable.
Todas las superficies del material deberán estar pulidas y exentas de grietas, y todos los ángulos redondeados. El equipo de toma de muestras deberá cumplir los requisitos establecidos con respecto a los productos de los que deban tomarse muestras.
3. Recipientes de toma de muestras
Características:
Los recipientes y cierres para muestras serán de unos materiales y una estructura que protejan convenientemente la muestra, y que no provoquen en ella cambios que puedan alterar el resultado de los análisis o exámenes. Entre los materiales idóneos se incluyen el vidrio, algunos metales y algunos plásticos. Los recipientes serán preferiblemente opacos. Si fuesen transparentes o translúcidos, los recipientes que contengan muestras deberán colocarse en lugar oscuro.
Los recipientes y sus cierres deberán hallarse limpios y secos.
La forma y la capacidad del recipiente será la idónea para satisfacer los requisitos establecidos para el producto del cual deba efectuarse una toma.
Podrán utilizarse recipientes de plástico de un solo uso, láminas de alumunio o bolsas de plástico adaptadas, dotados de sistemas de cierre apropiados.
Los recipientes distintos de las bolsas de plástico se cerrarán herméticamente, bien mediante un tapón idóneo o bien mediante un tapón a rosca de metal o plástico que posea, si ello es necesario; una junta estanca de plástico, insoluble, no absorbente e impermeable a las grasas, y que no altere el olor, el sabor, las propiedades o la composición de la muestra.
En caso de que se utilicen tapones, éstos deberán estar hechos o recubiertos de materiales no absorbentes e inodoros.
4. Técnicas de toma de muestras
El recipiente de toma de muestras se cerrará inmediatamente tras efectuarse la toma.
5. Almacenamiento de las muestras
Las temperaturas de almacenamiento de muestras recomendadas para las diversas caseínas y caseinatos no sobrepasarán los 25° C.
6. Transporte de muestras
Las muestras se remitirán al laboratorio responsable de las pruebas tan pronto como sea posible (preferentemente, antes de que transcurran veinticuatro horas tras la toma). Durante el transporte, deberán tomarse precauciones a fin de evitar que las muestras queden expuestas a olores contaminantes, luz solar directa y temperaturas superiores a 25° C.
II. MÉTODO: TOMA DE MUESTRAS DE CASEÍNAS Y CASEINATOS COMESTIBLES
1. Objeto y ámbito de aplicación
Este método describe la toma de muestras para el análisis químico de:
Caseínas ácidas comestibles.
Caseínas al cuajo comestibles.
Caseinatos comestibles.
2. Equipo
Véase la sección 2.ª de las disposiciones generales.
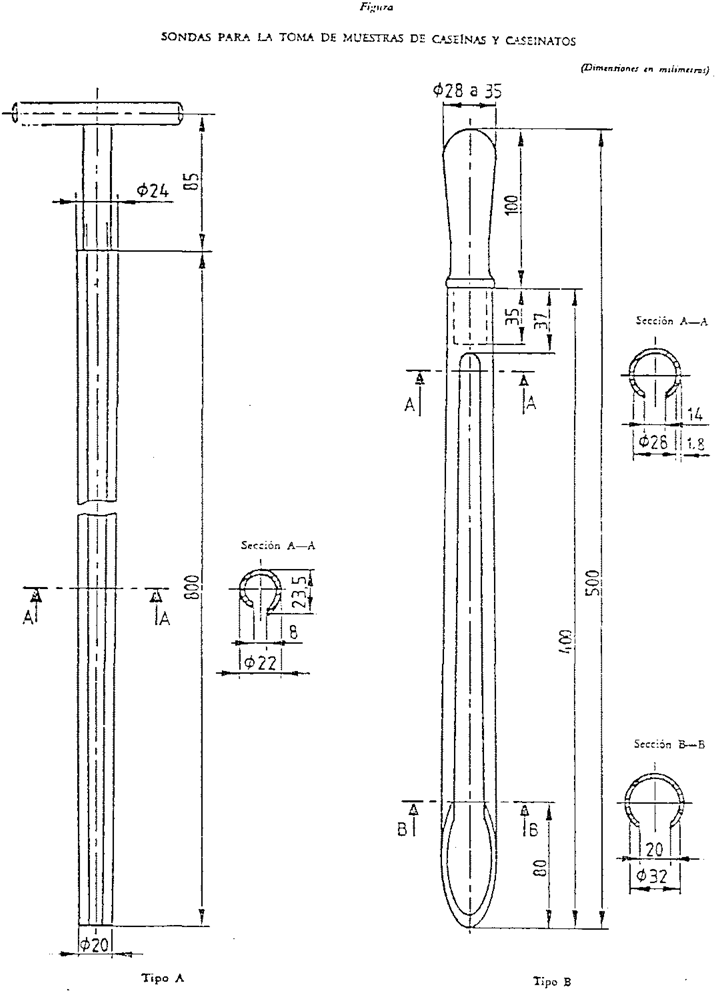
2.1 Sondas de longitud suficiente para alcanzar el fondo del recipiente del producto. Son idóneas las sondas correspondientes a la descripción que se da en la parte III del anexo 3 de esta Orden.
2.2 Cuchara, espátula o cucharilla. De hoja ancha.
2.3 Recipientes de muestra. Véase la sección 3 de las disposiciones generales.
3. Procedimiento
3.1 Generalidades: Se pondrá cuidado en minimizar la absorción de humedad atmosférica por el producto contenido en el recipiente durante o antes de la toma de muestra con fines analíticos. El recipiente se volverá a cerrar herméticamente una vez efectuada la toma de muestras.
3.2 Para el análisis químico.
3.2.1 Toma de muestras (en general). Se tomará una muestra no inferior a 200 gramos. Introducir en el producto una sonda limpia y seca, si es necesario con el recipiente inclinado o tumbado lateralmente. Se orientará la ranura hacia abajo y se adoptará un ritmo uniforme de penetración. Cuando la sonda alcance el fondo del recipiente se hará girar 180 grados, se extraerá y se descargará el contenido en el recipiente de la muestra. Para obtener una muestra no inferior a 200 gramos se efectuarán una o varias tomas. Se cerrará inmediatamente el recipiente una vez efectuada la toma. Esta toma se efectuará en el mismo lote o partida.
3.2.2 Toma de muestras de productos envasados en pequeños envases destinados a la venta al por menor.
La muestra podrá consistir en el contenido de un envase intacto y sin abrir. Siempre que sea posible, para constituir una muestra no inferior a 200 gramos se tomará uno o más envases de la misma partida o lote.
Si ello no fuera posible, utilícese otro método que permita obtener una muestra representativa.
3.2.3 Conservación, almacenamiento y transporte de muestras.
Véanse las secciones 5 y 6 de las disposiciones generales.
III. SONDAS PARA LA TOMA DE MUESTRAS DE CASEÍNAS Y CASEINATOS
1. Tipos de sondas
Tipo A: Larga (véase figura).
Tipo B: Corta (véase figura).
2. Materiales
La hoja y el eje deberán ser de metal pulido, preferiblemente de acero inoxidable.
El mango de tipo largo deberá ser preferiblemente de acero inoxidable.
La sonda de tipo corto poseerá un mango desmontable de madera o de plástico, fijado a la hoja con una unión a bayoneta.
3. Construcción
3.1 La forma, el material y el acabado deberán ser de tal manera que permitan fácilmente la limpieza.
3.2 El borde sobresaliente de la hoja del tipo A deberá ser lo suficientemente agudo, de forma que se pueda rascar con él.
3.3 La punta de la hoja deberá ser suficientemente aguda, de forma que facilite la toma de muestras.
4. Dimensiones principales
Las sondas deberán presentar las dimensiones que se dan en el cuadro inferior (con una tolerancia del 10 por 100).
|
|
Tipo A Larga (en mm) |
Tipo B Corta (en mm) |
|---|---|---|
|
Longitud de la hoja |
800 |
400 |
|
Espesor del metal de la hoja |
De 1 a 2 |
De 1 a 2 |
|
Diámetro interno de la hoja en la punta |
18 |
32 |
|
Diámetro interno de la hoja en el mango o el eje |
22 |
28 |
|
Anchura de la ranura en la punta |
4 |
20 |
|
Anchura de la ranura en el mango o el eje |
14 |
14 |
5. Advertencia sobre el uso de las sondas
5.1 Con polvo más o menos suelto, las sondas podrán introducirse verticalmente. Las sondas de tipo A se llenarán completamente haciéndose girar, y se extraerán a continuación verticalmente. Las sondas de tipo B se llenarán completamente a medida que se vayan introduciendo, pero deberán retirarse en posición oblicua a fin de evitar que se produzcan pérdidas por el extremo inferior.
5.2 En el caso de polvos sueltos deberán inclinarse los recipientes, introduciéndose las sondas en posición casi horizontal, con la ranura hacia abajo, para extraerse después con la ranura hacia arriba.
[Bloque 11: #anejo4]
DISPOSICIONES GENERALES
1. Preparación de la muestra de análisis
1.1 Observación general: La masa de la muestra presentada al laboratorio a efectos de su análisis debe ser de 200 gramos como mínimo.
1.2 Preparación de la muestra para el análisis en laboratorio.
1.2.1 Homogeneizar cuidadosamente la muestra de laboratorio, triturar los posibles grumos, sacudiendo y girando con frecuencia el recipiente (si es necesario, tras haber transferido toda la muestra a un recipiente hermético al aire de capacidad doble de volumen de la muestra a fin de poder efectuar esta operación).
1.2.2 Verter en el tamiz 3.3 una parte lo más representativa posible de la muestra cuidadosamente homogeneizada (alrededor de 50 gramos).
1.2.3 Si la totalidad de los 50 gramos atraviesa completa o casi completamente el tamiz (un 95 por 100 en peso, como mínimo), utilizar para la determinación de la muestra preparada según 1.2.1.
1.2.4 Si no es así, moler los 50 gramos empleando el dispositivo de moler (3.4) hasta que cumplan el criterio de tamizado (1.2.3). Pasar inmediatamente toda la muestra tamizada a un recipiente hermético al aire de capacidad doble del volumen de la muestra, y homogeneizar cuidadosamente agitando y girando continuamente. Durante estas operaciones, tomar precauciones para evitar toda modificación del contenido en agua del producto.
1.2.5 Tras preparar la muestra para la prueba, proceder todo lo rápidamente posible a la determinación.
1.3 Recipientes: La muestra debe mantenerse siempre en un recipiente hermético al aire y a la humedad.
2. Reactivos
2.1 Agua.
2.1.1 El agua utilizada para la preparación de solución, de dilución o de solución de lavado deberá ser agua destilada o agua desmineralizada de pureza al menos equivalente.
2.1.2 Cada vez que se haga referencia a una «solución» o a una «dilución» sin otra indicación, se tratará de una «solución acuosa» o de una «dilución agua».
2.2 Sustancias químicas: Salvo cuando se indique lo contrario, todas las sustancias químicas deberán ser de calidad analítica reconocida.
3. Material
3.1 Lista de material: Las listas de material sólo comprenderán artículos que tengan un empleo determinado y que respondan a características particulares.
3.2 Balanza analítica: Se entenderá por balanza analítica una balanza con una precisión de pesada de al menos 0,1 miligramos.
3.3 Tamiz: El tamiz deberá poseer un diámetro de 200 milímetros e ir provisto de una tapa y un recipiente colector. La tela metálica del tamiz ha de tener una abertura nominal de 500 µm. Las tolerancias de abertura y el diámetro del hilo metálico corresponderán a los valores indicados en la norma ISO/3310/1 (Tamiz para pruebas –normas técnicas y comprobación– primera parte: Tela metálica ISO/3310/1-1975, o la última edición de esta norma).
3.4 Dispositivo moledor: Permite, si es necesrio, moler la muestra de laboratorio sin provocar un calor excesivo y sin modificar la humedad (1.2.4). No deberá utilizarse un moledor de martillo.
4. Expresión de los resultados
4.1 Resultados: Si se cumplen las condiciones de reproducibilidad aplicables a un método, se tomará como resultado la media aritmética de dos determinaciones.
4.2 Cálculo de porcentaje: Salvo cuando se indique lo contrario, deberán calcularse los resultados como porcentaje en masa de la muestra.
5. Acta de la prueba
El acta de la prueba precisará el método analítico empleado y los resultados obtenidos. Incluirá también todos los detalles del procedimiento no previstos en el método analítico o facultativos, así como todas las circunstancias que hayan podido influenciar los resultados obtenidos. El acta de la prueba dará todas las informaciones necesarias para la identificación completa de la muestra.
MÉTODO 1
Determinación del contenido en humedad
1. Objeto y ámbito de aplicación
El presente método permite determinar el contenido en humedad de:
Las caseínas ácidas.
Las caseínas-cuajos.
Los caseinatos.
2. Definición
Por contenido en humedad de caseínas y caseinatos se entederá la pérdida de masa que se obtiene aplicando el método descrito a continuación.
3. Principio
Desecación a presión atmosférica, en una estufa a 102º C ± 1º C, de una muestra de prueba hasta obtener una masa constante, se calcula la pérdida de masa como porcentaje en masa de la muestra.
4. Equipo
4.1 Balanza analítica.
4.2 Cápsulas: De fondo plano y de material inalterable en las condiciones de la prueba (por ejemplo de niquel, aluminio, acero inoxidable o vidrio). Deben ir provistos de tapas que ajusten perfectamente pero que puedan retirarse rápidamente. Dimensiones adecuadas: Diámetro de 60 a 80 milímetros y profundidad de unos 20 milímetros.
4.3 Estufa a presión atmosférica: Bien ventilada y provista de un dispositivo de termorregulación (temperatura: 102º C ± 1º C). La temperatura ha de ser uniforme en todo el interior de la estufa.
4.4 Desecador: Deberá contener gel de sílice activado recientemente, provisto de un indicador hidrométrico, o un dispositivo de desecación equivalente.
4.5 Instrumento para manipular las cápsulas: Por ejemplo, pinzas de laboratorio.
5. Procedimiento
5.1 Preparación de la muestra para el ensayo: Véase el punto 1.2 de las disposiciones generales.
5.2 Preparación de las cápsulas.
5.2.1 Colocar la cápsula destapada y su tapa (4.2) en la estufa (4.3), regulada a 102º C ± 1º C, durante al menos una hora.
5.2.2 Colocar la tapa sobre la cápsula, introducir la cápsula así tapada en el desecador (4.4), dejar enfriar a temperatura ambiente y pesar con precisión de 0,1 miligramos (m0).
5.3 Muestra de prueba: Introducir de 3 a 5 gramos de la muestra para prueba (5.1) en la cápsula, tapar ésta y pesar con precisión de 0,1 miligramos (m1).
5.4 Determinación.
5.4.1 Destapar la cápsula y colocarla junto con la tapa en la estufa, regulada a 102º C ± 1º C; dejarla allí durante cuatro horas.
5.4.2 Colocar la tapa sobre la cápsula, introducirla en el desecador, dejar enfriar a temperatura ambiente y pesar con precisión de 0,1 miligramos.
5.4.3 Destapar la cápsula y colocarla junto con la tapa en la estufa durante una hora. Repetir a continuación la operación descrita en 5.4.2.
5.4.4 Si la masa obtenida según 5.4.3 es inferior en más de un miligramo a la obtenida según 5.4.2, repetir la operación descrita en 5.4.3.
Si se obtiene un aumento de la masa, utilizar para el cálculo la cifra más baja registrada (6.1).
La duración total del secado no sebrepasa por lo general seis horas.
Designemos como m2 la masa final registrada, en gramos.
6. Expresión de los resultados
6.1 Método de cálculo: La pérdida de masa de la muestra de prueba, expresada en porcentaje de masa, viene dado por la fórmula siguiente:
|
|
μ1 – μ2 |
× 100 |
|
|
μ1 – μ0 |
donde:
m0 = Masa en gramos de la cápsula y su tapa tras la operación 5.2.
m1 = Masa en gramos de la cápsula, su tapa y la muestra de prueba antes del secado (5.3).
m2 = Masa en gramos de la cápsula, su tapa y la muestra de prueba tras el secado (5.4.3 ó 5.4.4).
Expresar el resultado final con precisión de 0,01 por 110.
6.2 Reproducibilidad: La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente con un breve intervalo sobre la misma muestra, en las mismas condiciones y por el mismo analista, no debe sobrepasar los 0,1 gramos de humedad por 100 gramos de producto.
La probabilidad de no sobrepasar este valor es del 95 por 100 de los casos.
MÉTODO 2
Determinación del contenido en proteínas
1. Objeto y ámbito de aplicación
El presente método permite determinar el contenido en proteínas de:
Las caseínas ácidas, las caseínas-cuajos y los caseinatos.
Con excepción de los caseinatos que contengan amonio y otros compuestos de amonio o nitrógeno no proteico.
2. Definición
Contenido en proteínas: Contenido en nitrógeno determinado por el método aquí descrito, multiplicado por 6,38 y expresado como porcentaje en masa.
3. Principio
Se ataca una muestra de prueba con una mezcla de sulfato de potasio y de ácido sulfúrico en presencia de sulfato de cobre (II) como catalizador, a fin de transformar el nitrógeno orgánico en nitrógeno amoniacal. Se destila el amoníaco y se absorbe en una solución de ácido bórico.
Se valora mediante una solución estándar de ácido clorhídrico. Se obtiene el contenido en proteínas multiplicando el contenido en nitrógeno por 6,38.
4. Reactivos
4.1 Ácido sulfúrico concentrado D20, 1,84 g/ml.
4.2 Sulfato de potasio anhidro (K2SO4).
4.3 Sulfato de cobre (II) pentahidratado (CuSO4-5H2O).
4.4 Sacarosa (C12H22O11).
4.5 Ácido bórico, solución de 40 g/l.
4.6 Hidróxido sódico, solución concentrada al 30 por 100 en masa, exenta de carbonato.
4.7 Ácido clorhídrico: Solución estándar a 0,1 mol/l.
4.8 Indicador mixto: Mezclar a volúmenes iguales una solución de rojo de metilo de 2 g/l en etanol de, al menos, 95 por 100 (en volumen), y una solución de azul de metileno de 1 g/l en etanol de, al menos, 95 por 100 (en volumen).
5. Equipo
5.1 Balanza analítica.
5.2 Matraz de Kjeldahl, 500 mililitros.
5.3 Aparato de descomposición proteica. Debe permitir mantener inclinado el matraz de Kjeldahl (5.2) e ir provisto de un sistema de calentamiento que evite el calentamiento de la parte del matraz situada por encima del nivel del líquido.
5.4 Condensador de tubo interior rectilíneo.
5.5 Tubo de salida. Con una ampolla de seguridad unida al extremo inferior del condensador (5.4) por una conexión en vidrio esmerilado o un tubo de goma. En este último caso, los extremos de vidrio han de hallarse próximos entre sí.
5.6 Deflegmador. Unido al matraz de Kjeldahl (5.2) y al condensador (5.4) mediante tapones de goma bien ajustados.
5.7 Matraz Erlenmeyer, 500 mililitros.
5.8 Probetas graduadas, 50 mililitros y 100 mililitros.
5.9 Bureta de 50 mililitros, graduaciones a 0,1 mililitro.
5.10 Reguladores de ebullición.
5.10.1 Para la descomposición proteica: Trozos pequeños de porcelana o bolas de vidrio.
5.10.2 Para la destilación: Gránulos pequeños de piedra pómez.
6. Procedimiento
6.1 Preparación de la muestra para la prueba. Véase el punto 1.2 de las disposiciones generales.
6.2 Demostración del nitrógeno amoniacal. Si se supone la presencia de caseinato de amonio o de otros compuestos amoniacales, efectuar la prueba siguiente: En un pequeño matraz Erlenmeyer, añadir a 1 gramo de muestra 10 mililitros de agua y 100 miligramos de óxido de magnesio. Enjuagar el óxido adherido a la pared de vidrio, tapar el matraz con un tapón de corcho, metiendo entre el tapón y el cuello del matraz una tira de papel tornasol humectada. Mezclar con cuidado el contenido del matraz y calentar éste en un baño a unos 60º C. Si el papel tornasol vira a azul en los quince minutos siguientes, esto revela la presencia de amoníaco; en este caso, el método no es aplicable (véase el punto 1).
6.3 Prueba en blanco. Simultáneamente a la determinación del contenido en nitrógeno de la prueba, efectuar una prueba en blanco sustituyendo la muestra de prueba por 0,5 gramos de sacarosa (4.4) y utilizando el mismo equipo, las mismas cantidades de reactivo y el mismo procedimiento que los descritos en el punto 6.5. Si en la prueba en blanco la valoración sobrepasa 0,5 mililitros de la solución ácida de 0,1 mol/l, deberán verificarse los reactivos y purificarse o sustituirse el reactivo o los reactivos impuros.
6.4 Muestra de prueba. Introducir en el matraz de Kjeldahl (5.2) de 0,3 a 0,4 gramos de la muestra (6.1), pesados con precisión de 0,1 miligramos.
6.5 Determinación.
6.5.1 Introducir en el matraz algunos trozos de porcelana o algunas bolas de vidrio (5.10.1) y aproximadamente 10 gramos de sulfato de potasio anhidro (4.2).
Añadir 0,2 gramos de sulfato de cobre (II) (4.3) y enjuagar el cuello del matraz con un poco de agua. Añadir 20 mililitros de ácido sulfúrico concentrado (4.1) y mezclar el contenido del matraz.
Calentar suavemente sobre el aparato de descomposición (5.3) hasta que desaparezca la espuma. Hacer ebullir suavemente hasta que la solución esté límpida y persista el color verdiazul pálido. Agitar el matraz de tiempo en tiempo durante el calentamiento.
Proseguir la ebullición regulando el calentamiento, de forma que los vapores se condensen en el centro del cuello del matraz. Continuar calentando durante noventa minutos, evitando todo sobre calentamiento local.
Dejar enfriar a temperatura ambiente. Añadir con precaución unos 200 mililitros de agua y algunos granos de piedra pómez (5.10.2). Mezclar y dejar enfriar de nuevo.
6.5.2 Introducir en el matraz Erlenmeyer (5.7) 50 mililitros de la solución de ácido bórico (4.5) y cuatro gotas del indicador (4.8). Mezclar. Colocar el matraz Erlenmeyer bajo el condensador (5.4) de manera que la extremidad del tubo de salida (5.5) esté sumergida en la solución de ácido bórico. Mediante una probeta graduada (5.8) introducir en el matraz de Kjeldahl 80 mililitros de la solución de hidróxido sódico (4.6). Durante esta operación debe mantenerse el matraz inclinado de forma que la solución de hidróxido de sodio fluya a lo largo de la pared y forme una capa en la parte inferior del matraz.
Volver a acoplar inmediatamente el matraz de Kjeldahl al condensador por medio del deflegmador (5.6).
Mezclar el contenido del matraz de Kjeldahl haciéndolo girar suavemente, hacer ebullir suavemente al principio, evitando toda formación de espuma. Continuar la destilación de forma que se obtengan 150 mililitros de destilado en treinta minutos, aproximadamente.
El destilado debe poseer una temperatura inferior a 25º C.
Unos dos minutos tras el término de la destilación, bajar el matraz Erlenmeyer de manera que el extremo del tubo de salida no esté ya sumergido en la solución ácida, y enjuagar dicho extremo con un poco de agua. Hacer cesar el calentamiento, levantar el tubo de salida y enjuagar sus paredes interiores y exteriores con un poco de agua, recogiendo el agua de enjuagado en el matraz Erlenmeyer.
6.5.3 Valorar el destilado del matraz Erlenmeyer mediante la solución estándar de ácido clorhídrico (4.7).
7. Expresión de los resultados
7.1 Modo de cálculo y fórmula. El contenido en proteínas de la muestra, expresado en porcentaje en masa, es igual a:
|
(V1 – V2) × T × 14 × 100 × 6,38 |
= |
8,932 (V1 – V2) × T |
|
m × 1.000 |
m |
donde:
V1 = Volumen en mililitros de la solución estándar de ácido clorhídrico (4.7) utilizado para la determinación (6.5).
V2 = Volumen en mililitros de la solución estándar de ácido clorhídrico (4.7) utilizada para el ensayo en blanco.
T = Concentración de la solución estándar de ácido clorhídrico (4.7) en mol/l.
m = Masa, en gramo de la muestra de prueba.
Expresar el contenido en proteínas con precisión de 0,1 por 100.
7.2 Reproducibilidad. La diferencia entre los resultados de dos determinaciones efectuadas sumultáneamente con un breve intervalo, en las mismas condiciones y por el mismo analista, no debe sobrepasar los 0,5 gramos de proteínas por 100 gramos de producto.
Este valor tiene una probabilidad de no sobrepasarse del 95 por 100 de los casos, si el método se lleva a cabo de forma correcta.
MÉTODO 3
Determinación de la acidez valorable
1. Objeto y ámbito de aplicación
El presente método permite determinar la acidez valorable de las caseínas ácidas.
2. Definición
Acidez valorable de las caseínas ácidas: Volumen, en mililitros de solución de hidróxido sódico de 0,1 mol/l, necesario para neutralizar un extracto acuoso de un gramo de producto.
3. Principio
Preparación y filtración de un extracto acuoso de la muestra a 60º C. Valoración del filtrado mediante una solución estándar de hidróxido sódico, en presencia de fenolftaleína como indicador.
4. Reactivos
El agua utilizada para la aplicación del método o la preparación de los reactivos debe estar exenta de anhídrido carbónico (a tal fin, calentarla diez minutos antes de usarla).
4.1 Hidróxido sódico, solución a 0,1 mol/l.
4.2 Fenolftaleína utilizada como indicador, solución de un gramo en 100 mililitros de etanol (95 por 100 en volumen), neutralizada con relación a un indicador.
5. Equipo
5.1 Balanza analítica.
5.2 Matraz Erlenmeyer, 500 mililitros, con cuello y tapón esmerilados.
5.3 Pipeta graduada a 100 mililitros.
5.4 Pipeta, adecuada para medir 0,5 mililitros de la solución indicadora (4.2).
5.5 Matraz Erlenmeyer, 250 mililitros.
5.6 Probeta graduada, 250 mililitros.
5.7 Bureta con graduaciones de 0,1 mililitros.
5.8 Baño de agua, regulable a una temperatura de 60 ± 2º C.
5.9 Filtro adecuado.
6. Procedimiento
6.1 Para la preparación de la muestra para la prueba, véase el punto 1.2 de las disposiciones generales.
6.2 Muestra de prueba: Pesar unos diez gramos de la muestra con precisión de 10 miligramos e introducirlos en el matraz Erlenmeyer (5.2).
6.3 Determinación: Mediante la probeta de 250 mililitros (5.6), añadir 200 mililitros de agua recientemente ebullida y enfriada, calentada previamente a 60º C. Cerrar el matraz, mezclar por agitación de éste y colocarlo en el baño a 60º C (5.8) durante treinta minutos. Agitar el matraz cada diez minutos, aproximadamente.
Filtrar y dejar enfriar el filtrado a unos 20º C. El filtrado debe ser límpido.
Tomar con la pipeta (5.3) 100 mililitros del filtrado enfriado e introducirlos en el matraz Erlenmeyer (5.5). Añadir 0,5 mililitros de la solución indicadora de fenolftaleína (4.2) mediante la pipeta (5.4). Valorar mediante la solución estándar de hidróxido sódico (4.1) hasta que aparezca un color rosa pálido persistente durante al menos treinta segundos. Anotar el volumen utilizado con precisión de 0,01 mililitros.
7. Expresión de los resultados
7.1 La acidez valorable de la caseína viene dada por la fórmula:
|
|
20 × V × T |
|
|
|
m |
|
donde
V = Volumen en mililitros de la solución estándar de hidróxido sódico (4.1) utilizada.
T = Concentración, en mol/l, de la solución estándar de hidróxido sódico (4.1).
m = Masa, en gramos, de la muestra de prueba.
Expresar la acidez valorable con dos decimales.
7.2 Reproducibilidad: La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o con un breve intervalo, sobre la misma muestra, en las mismas condiciones y por el mismo analista, no debe sobrepasar 0,02 mililitros de solución de hidróxido sódico para un gramo de producto.
Este valor tiene una probabilidad de no ser sobrepasado del 95 por 100 de los casos.
MÉTODO 4
Determinación de cenizas (P2 O5 incluido)
1. Objeto y ámbito de aplicación
El presente método permite determinar las cenizas de: Las caseínas ácidas (P2 O5 incluido).
2. Definición
Cenizas (P2O5 incluido): Sustancias determinadas según el metodo descrito a continuación, expresadas como porcentaje en masa.
3. Principio
Incineración de una muestra de prueba a 825º C ± 25º C en presencia de acetato de magnesio, destinado a fijar la totalidad del fósforo de origen orgánico. Se pesa el residuo y se sustrae la masa de cenizas procedentes del acetato de magnesio.
4. Reactivos
4.1 Acetato de magnesio tetrahidratado
Mg (CH3CO2)2, 4H2O, solución de 120 g/l
5. Equipo
5.1 Balanza analítica.
5.2 Pipeta graduada a 5 mililitros.
5.3 Cápsula de silicio o platino, de unos 70 milímetros de diámetro y de 25 a 50 milímetros de profundidad.
5.4 Estufa de secado regulable a 102º C ± 1º C.
5.5 Horno eléctrico de circulación de aire, regulable a 825º C ± 25º C.
5.6 Baño de agua en ebullición.
5.7 Desecador con gel de sílice recientemente activado, provisto de un indicador hidrométrico, o un deshidratante equivalente.
6. Procedimiento
6.1 Preparación de la muestra para la prueba: Véase el punto 1.2 de las disposiciones generales.
6.2 Preparación de las cápsulas: Colocar dos cápsulas (5.3) A y B en el horno eléctrico (5.5), regulado a 825º C ± 25º C, durante treinta minutos. Dejarlas enfriar un poco antes de colocarlas en un desecador (5.7) hasta que se enfríen a la temperatura ambiente, y pesarlas con precisión de 0,1 miligramo.
6.3 Muestra de prueba: Pesar con precisión de 0,1 miligramo unos tres gramos de la muestra (6.1), directamente sobre una de las cápsulas preparadas (A).
6.4 Determinación: Introducir en la cápsula A, mediante la pipeta (5.2), cinco mililitros exactos de la solución de acetato de magnesio (4.1), de manera que se humedezca toda la muestra de prueba, y dejar reposar durante veinte minutos.
En la otra cápsula preparada (B), introducir con la pipeta (5.2) 5 mililitros exactos de la solución de acetato de magnesio (4.1).
Evaporar hasta la sequedad el contenido de ambas cápsulas A y B, en el baño de agua en ebullición (5.6).
Colocar las dos cápsulas en la estufa (5.4), regulada a 102º C ± 1º C, durante treinta minutos.
Colocar la cápsula A, con su contenido, sobre una llama pequeña, una placa caliente o bajo una lámpara de infrarrojos, hasta que la muestra esté completamente carbonizada y vigilando que ésta no arda.
Colocar las dos cápsulas A y B en un horno eléctrico (5.5) regulado a 825º C ± 25º C, y dejarlas allí durante al menos una hora, hasta que las partículas carbonosas de la cápsula A desaparezcan completamente, dejar enfriar un poco ambas cápsulas y colocarlas en el desecador (5.7) hasta que alcancen la temperatura ambiente, pesándolas luego con precisión de 0,1 miligramos.
Repetir las operaciones de calentamiento durante unos treinta minutos en el horno eléctrico, de enfriamiento y de pesada hasta obtener una masa constante (con precisión de un miligramo), anotando la masa mínima.
7. Expresión de los resultados
7.1 Modo y fórmula de cálculo: Las cenizas (P2O5 incluido) de la muestra, expresadas como porcentaje en masa, viene dadas por la fórmula:
|
|
(m1 – m2) – (m3 – m4) |
× 100 |
|
|
m0 |
donde
m0 = Masa, en gramos, de la muestra de prueba.
m1= Masa, en gramos, de la cápsula A con su contenido.
m2 = Masa, en gramos, de la cápsula A vacía.
m3 = Masa, en gramos, de la cápsula B con su contenido.
m4 = Masa, en gramos, de la cápsula B vacía.
Expresar el resultado final con precisión de 0,01 por 100.
7.2 Reproducibilidad: la diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o con un breve intervalo sobre la misma prueba, en las mismas condiciones y por el mismo analista, no debe sobrepasar los 0,1 gramos por 100 gramos de producto.
Este valor tiene una probabilidad de no ser sobrepasado del 95 por 100 de los casos.
MÉTODO 5
Determinación de cenizas (P2O5 incluido)
1. Objeto y ámbito de aplicación
El presente método permite determinar el contenido en cenizas de las caseínas puras.
2. Definición
Cenizas (P2O5 incluido): Sustancias determinadas según el método descrito a continuación y expresados como porcentaje en masa.
3. Principio
Incineración de una muestra de prueba a 825º C ± 25º C hasta la obtención de una masa constante. Se pesa el residuo obtenido.
4. Equipo
4.1 Balanza analítica.
4.2 Cápsula de silicio o platino, de aproximadamente 70 mm de diámetro y de 25 a 50 mm de profundidad.
4.3 Horno eléctrico. Horno de circulación de aire, regulable a 825º C± 25º C.
4.4 Desecador. Con gel de sílice activado recientemente, provisto de un indicador hidrométrico, o cualquier deshidratante equivalente.
5. Procedimiento
5.1 Preparación de la muestra para la prueba. Véase el punto 1.2 de las «Disposiciones generales».
5.2 Preparación de la cápsula. Colocar la cápsula (4.2) en el horno eléctrico (4.3), regulado a 825º C ± 25º C, durante treinta minutos. Dejar enfriar la cápsula en el desecador (4.4) hasta la temperatura ambiente. Pesar con precisión de 0,1 mg.
5.3 Muestra de prueba. Pesar directamente en la cápsula preparada, con precisión de 0,1 mg, unos 3 g de la muestra (5.1).
5.4 Deteminacion. Calentar la cápsula, con su contenido, sobre una llama pequeña, una placa caliente o bajo una lámpara de infrarrojos, hasta que la muestra esté completamente carbonizada y vigilando que ésta no arda.
Colocar la cápsula en un horno eléctrico (4.3) regulado a 825º C ± 25º C y dejarla allí durante al menos una hora, hasta que las partículas carbonosas de la cápsula desaparezcan completamente. Dejar enfriar un poco la cápsula y colocarla en el desecador (4.4) hasta que alcance la temperatura ambiente, pesándola luego con precisión de 0,1 mg.
Repetir las operaciones de calentamiento durante unos treinta minutos en el horno eléctrico, de enfriamiento y de pesada hasta obtener una masa constante (con precisión de 1 mg).
6. Expresión de los resultados
6.1 Método y fórmula de cálculo. Las cenizas (P2O5 incluido) de la muestra, expresadas como porcentaje en masa, vienen dadas por la fórmula:
|
|
m1 – m2 |
× 100 |
|
|
m0 |
donde
m0 = Masa, en gramos, de la muestra de prueba.
m1 = Masa, en gramos, de la cápsula con su contenido.
m2 = Masa, en gramos, de la cápsula vacía.
Expresar el resultado final con precisión de 0,01 por 100.
6.2 Reproducibilidad. La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o con un breve intervalo sobre la misma prueba, en las mismas condiciones y por el mismo analista, no debe sobrepasar 0,15 g de cenizas por 100 g de producto.
Este valor tiene una probabilidad de no ser sobrepasado del 95 por 100 de los casos.
MÉTODO 6
Determinación del pH
1. Objeto y ámbito de aplicación
El presente método permite determinar el pH de los caseinatos.
2. Definición
pH de los caseinatos: El pH a 20º C de una solución de caseinato, determinado según el método descrito a continuación.
3. Principio
Determinación electrométrica del pH de una solución de caseinato mediante un pHmetro.
4. Reactivos
El agua utilizada para la preparación de reactivos o para la realización del método (6) debe ser agua recientemente destilada y protegida contra la absorción de anhídrido carbónico.
4.1 Soluciones tampón para la calibración del Phmetro. Dos soluciones tampón patrones, de un pH dado a 20º C con precisión de dos cifras decimales y cuyo intervalo cubra el pH de la muestra; por ejemplo, una solución tampón de ftalato, de pH próximo a 4, y una solución tampón de bórax, de pH próximo a 9.
5. Equipo
5.1 Balanza, sensibilidad de 0,1 g.
5.2 pHmetro. Sensibilidad mínima: 0,5 pH, provisto de un electrodo de vidrio adecuado y de un electrodo de calomel u otro electrodo de referencia.
5.3 Termómetro. Precisión: 0,5º C.
5.4 Matraz Erlenmeyer. Capacidad de 100 ml, provisto de un tapón de vidrio esmerilado.
5.5 Vaso de vidrio de 50 ml.
5.6 Agitador.
5.7 Vaso para el agitador (5.6), capacidad mínima 250 ml.
6. Procedimiento
6.1 Preparación de la muestra para la prueba. Véase el punto 1.2 de las «disposiciones generales».
6.2 Determinación.
6.2.1 Calibración del pHmetro. Establecer la temperatura de las soluciones tampón (4.1) en 20º C y regular el pHmetro según las instrucciones del fabricante.
Observaciones:
6.2.1.1 El calibrado debe efectuarse introduciendo el electrodo durante veinte minutos en los matraces Erlenmeyer en reposo (6.2.2).
6.2.1.2 Si se efectúa el análisis de una serie de muestras, verificar el calibrado del pH al menos cada treinta minutos, con una o varias soluciones tampón patrones.
6.2.2 Preparación de la solución de prueba. Introducir en el vaso de vidrio (5.6) 95 ml de agua, añadir 5,0 g de la muestra de prueba (6.1) y mezclar mediante el agitador (5.5) durante treinta segundos.
Dejar reposar durante veinte minutos a unos 20º C, con el vaso tapado con un vidrio de reloj.
6.2.3 Medida del pH.
6.2.3.1 Verter unos 20 ml de la solución en el vaso (5.5) y determinar inmediatamente el pH de este líquido con el pHmetro (5.2), tras haber enjuagado cuidadosamente los electrodos con agua.
6.2.3.2 Medir el pH.
7. Expresión de los resultados
7.1 Lectura del pH. Anotar el valor leído sobre la escala del pHmetro que corresponda al pH de la solución de caseinato, con dos cifras decimales como mínimo.
7.2 Repetibilidad. La diferencia entre los resultados de dos determinaciones efectuadas simultáneamente o con un breve intervalo sobre la misma prueba, en las mismas condiciones y por el mismo analista, no debe sobrepasar los 0,05 unidades de pH.
Este valor tiene un probabilidad de no ser sobrepasado del 95 por 100 de los casos.
Este documento es de carácter informativo y no tiene valor jurídico.
Ayúdenos a mejorar: puede dirigir sus comentarios y sugerencias a nuestro Servicio de atención al ciudadano
Agencia Estatal Boletín Oficial del Estado
Avda. de Manoteras, 54 - 28050 Madrid





